

 ホーム
ホーム 最新情報
最新情報 機構紹介
機構紹介 国際交流
国際交流 サイトについて
サイトについて2020年 7月 2日医薬品審査センターは「国家医薬品監督管理局による『医薬品登録管理規則』実施関係事項の公告」(2020年第 46号)に基づき、関連付属規範性文書、技術ガイドラインの起草と制定業務を推進するために、国家医薬品監督管理局の審査をへて同意を得た上で「生物製品登録受理と審査マニュアル(第一部 予防用生物製品)」、「生物製品登録受理と審査マニュアル(第二部 治療用生物製品)」及び「生物製品登録受理と審査マニュアル(第三部 生物製品として管理される体外診断用試薬)」を発布し、発布日から施行する。
別添:
1、生物製品登録受理と審査マニュアル(第一部 予防用生物製品)(試行版)
2、生物製品登録受理と審査マニュアル(第二部 治療用生物製品)(試行版)
3、生物製品登録受理と審査マニュアル(第三部 生物製品として管理される体外診断用試薬)(試行版)
別添
生物製品登録受理審査マニュアル(試行版)
第一部 予防用生物製品
本マニュアルが現行の法律、法規の要求に基づいて制定する。本マニュアルで言及されず、または明確に規定されていない受理関係事項については、申請者が受理機関と意思疎通を行うことができる。今後、本マニュアルが関連法律、法規などの書類の要求に基づいて随時更新する。
一、適用範囲
予防用生物製品の臨床試験申請または上市許可申請。予防用生物製品とは、疾病の発生と流行を予防、コントロールするために、人体の免疫接種に用いるワクチン類生物製品を指し、国家免疫計画に係わるワクチンとそれ以外のワクチンを含む。
二、受理機関
国家医薬品監督管理局医薬品審査センターが受理する。
三、資料に関する基本的な要求
「医薬品登録管理規則」及び「生物製品登録分類及び申請資料要求」の規定に基づき、要求に適合する申請資料を提供する。申請資料は「M4:ヒト用医薬品承認申請のための国際共通化資料(CTD))」(以下「CTD」と略称)に基づいてまとめなければならない。また、目次及び項目番号は変更してはならず、関連情報または研究資料がない項目があっても、その番号及び名称を保留すべきで、その下に「不適用」と明記し、理由を説明する。
(一)申請表のまとめ
医薬品登録申請表、申請資料自主チェック表、小型・零細企業向け費用徴収優遇政策利用申請表(適用する場合のみ)は申請資料の部数と一致し、少なくともそのうちの1部は原本である。記入は正確、完全、規範的でなければならず、手書きまたは修正が禁止で、且つ申請表記入に関する説明の要求に適合していなければならない。
新版医薬品登録申請表作成・提出プログラムの使用に関する公告に基づき、申請表の記入と提出は国家医薬品監督管理局の統一的にリリースしたソフトウェアを使用し、新版「医薬品登録申請表作成・提出プログラム」で作成した電子版及び紙版の書類を提出しなければならない。使用中のバージョンが最新バージョン(最新の公告に準じる)であること及び生成される電子ファイルがRVT様式であることを確認しなければならない。各ページのデータ照合コードは一致し、且つ提出された電子版申請表と一致していなければならない。申請表及び自主チェック表の各ページの端には、申請者または登録代理機構の割印を押さなければならない。
(二)申請資料の整理
2セットの完全な申請資料(少なくとも1セットは原本)+lセットの総括資料(様式1、様式2を含まなければならない)。各セットに関連申請表及び目次を入れる。
「医薬品登録申請表」及び検定機関が作成する検定報告書を除き、申請資料(クロマトグラフを含む)は各表紙に申請者または登録代理機構の印鑑を押捺し、且つ文字のあるところに押印しなければならない。資料整理と整理要求の詳細は「医薬品登録申請資料様式、体裁及び整理規範」を参照するように。
四、形式審査の要点
(一)申請事項審査の要点
1、医薬品臨床試験終了後、医薬品臨床試験を再開して継続しようとする場合、改めて医薬品臨床試験申請を行わなければならない。医薬品臨床試験申請が認可された日から3年以内に、被験者が説明同意書に署名していない場合、当該医薬品臨床試験許可が自動的に失効する。医薬品臨床試験を実施する必要はまだある場合、改めて申請しなければならない。
2、ワクチン上市許可申請の審査期間中、ワクチンの安全性、有効性及び品質制御可能性に影響を及ぼす可能性のある重大変更が発生した場合、申請者は元の登録申請を取り下げ、補充研究後に改めて申請しなければならない。申請者の名称変更、登録住所名称変更等、技術審査評価内容に係わらない場合、速やかに書面で医薬品審査センターに告知し、証明資料を提出しなければならない。
(二)意思疎通交流に関する審査の要点
1、申請者が初回新薬臨床試験申請を行う前に医薬品審査センターに意思疎通交流会議の開催を申し込まなければならない。
2、上市許可申請を行う前に、申請者は医薬品審査センターに意思疎通会議の開催を申し込まなければならない。
3、意思疎通交流は「医薬品研究開発と技術審査関連意思疎通交流管理規則」の規定に適合しなければならない。
4、意思疎通交流を申し込んだ場合、申請時に発行された意思疎通交流番号、意思疎通に関する回答意見書を提出し、意見書における質問項目に各項目ごとに回答しなければならない。
(三)申請表審査の要点![]()
1、医薬品登録申請表
申請表は医薬品登録申請表記入説明の要求規範に基づいて記入されなければならず、記入した情報は証明書類における関連内容と一致しなければならない。
1.1、ワクチン上市登録プロセスの加速について。当該申請の実際状況に基づいて選択肢にチェックを入れる。意思疎通交流をへて確認した後、「優先審査承認手順」を選択した場合には、ワクチン上市許可申請を行うと同時に、「優先審査承認業務手順」に従って優先審査承認を申請する。「特別審査承認手順」を選択した場合には、「医薬品特別審査承認手順」に従って実施しなければならない。
1.2、申請事項について。当該申請の実際の申請事項に基づいて記入する。臨床研究を申請する場合、臨床試験を選択する。上市を申請する場合、上市許可を選択する。
1.3、ワクチン登録分類について。「生物製品登録分類及び申請資料要求」における予防用生物製品関連要求に従って登録分類を明確化しなければならない。
1.4、その他の事項について。小型・零細企業の条件に合致する企業が費用徴収優遇政策利用を申請する場合、小型・零細企業優遇を選択できる。
1.5、医薬品一般名称について。国家医薬品基準または医薬品登録基準に収録された医薬品一般名称を使用しなければならない。国家医薬品基準または医薬品登録基準に収録されていない名称を使用する場合、申請者は医薬品上市許可申請を行う際に一般名称証明書類の原本を提出し、若しくは同時に一般名称認定申請を行わなければならない。
1.6、同一品目の受理済または同時申請したその他の製剤及び規格について。当該品目の受理済または同時申請した製剤、あるいは規格が異なる品目の受理番号及び名称を記入する。臨床研究を完了し、上市を申請する場合には、元の臨床申請受理番号、臨床試験承認番号、臨床試験登録番号を記入しなければならない。![]()
1.7、原薬、添加剤と包装資材の由来について。ワクチン登録申請を行う際には、使用する添加剤、包装資材に関する情報を記入しなければならず、且つ記入した情報が提出した証明書類または医薬品用添加剤と包装資材登記情報公示プラットフォームに登記した相応内容と一致しなければならない。
1.8、主要適応症または主要治療効能について。主要効能と用途を簡潔に記入し、申請資料における予防可能疾患の情報をすべて含まなければならない。
1.9、今回の申請の回数について。関連品目の申請は今回で何回目であるかを記入する。過去の申請及び審査承認状況を簡単に説明する。例えば、申請者が自ら撤回し、または資料が審査承認の要求に適合しないため、国家医薬品監督管理局に承認されなかった等の状況。元の申請の審査承認が終了した後、再度申請できる。
1.10、申請者及び委託研究機構
記入された情報は証明書類と申請資料と一致しなければならない。そのうちの一つの申請機構を国に登録費用を納付する業務の担当者として指定する。申請者として記入された各機構は、いずれもその法定代表者またはその授権を受けた者(署名済授権書の原本を別途提供する必要がある)が署名し、機構の公印(機構名称と完全に一致しなければならない)を押捺しなければならない。
2、小型・零細企業向け費用徴収優遇政策利用申請表
小型・零細企業向け行政事業費用徴収優遇政策の利用条件に合致する場合、小型・零細企業向け費用徴収優遇政策利用申請表を提出し、以下の情報も提供する。
2.1、基本情報。企業名称、連絡担当者、連絡用電話番号等。いずれも「医薬品登録申請表」の関連情報と一致しなければならない。![]()
2.2、従業員、前納税年度の営業収入、企業資産総額等。申請者が実際の状況に基づいて記入する。
2.3、その法定代表者またはその授権を受けた者(署名済授権書の原本を別途提供する必要がある)が署名し、機構の公印(機構名称と完全に一致しなければならない)を押捺しなければならない。
(四)申請資料審査の要点国家医薬品監管機関の発表した証明事項取消し公告に「内部確認審査へ変更」と規定された証明事項については、公告の要求に準じる。
1、製品関連証明書類
1.1、医薬品用添加剤及び包装資材の証明書類。
1.1.1、医薬品用添加剤及び包装資材の合法的な由来に関する証明書類。供給契約書、領収書等が含まれなければならない(登録済原薬、医薬品用添加剤及び包装資材を選択して使用しない製剤の場合に適用する)。
1.1.2、医薬品用添加剤及び包装資材使用授権書の写し(製剤が登記済原薬、医薬品用添加剤及び包装資材を選択して使用する場合に適用する)。サプライヤーが当該書類を提供する場合、原薬、医薬品用添加剤と包装資材生産企業の授権が必要で、授権書類のコピーを添付しなければならない。
1.2、特許情報及び証明書類
申請対象となる医薬品または使用する処方箋、生産技術と手順、用途などに関する特許状況及びその権利帰属状況の説明文、並びに他人の特許権を侵害しない旨の誓約書。申請者が作成し、権利侵害の可能性がある場合には、全責任を負うと約束しなければならない。
1.3、対照医薬品由来を証明する書類(該当する場合のみ)。
1.4、商標情報及び証明書類(該当する場合のみ)。
商品名の使用を申請する場合、商標登録証を提供しなければならない。
1.5、医薬品臨床試験に関する証明書類(上市許可申請に適用する)![]()
「医薬品臨床試験承認書」または臨床試験通知書のコピー及び臨床試験実施プロセスにおける関連補充申請書類のコピー(あれば、提出する)、臨床試験用医薬品品質基準及び臨床試験登録番号等の関連資料を提出し、並びに「医薬品臨床試験承認書」または臨床試験通知書における意見について各項目ごとに回答する。
1.6、研究機構資格の証明書類
非臨床研究安全性評価機構は、医薬品監督管理機関の発行し、「医薬品非臨床研究品質管理規範」(略称はGLP)に適合する承認証明書類または検査報告書等の証明書類を提供しなければならない。臨床試験機構は届出証明書類を提供しなければならない。
1.7、医薬品医療機器コンビネーション製品に関する証明書類
属性判定により、医薬品に属し、または医薬品をメインとする医薬品医療機械コンビネーション製品であると確認された場合、医薬品医療機器コンビネーション製品属性判定結果通知書の写しを提出しなければならない。
1.8、臨床試験承認証明書類
海外未上市のワクチンは、海外医薬品管理機関の発行したワクチン臨床試験承認証明書類(あれば、提出する)またはその他の関連証明書類、公証認証文書及びその中国語訳を提出しなければならない。
1.9、ワクチン上市販売許可証明書類
海外上市済のワクチンは、海外医薬品管理機関が発行したワクチン上市販売許可書類、公証認証文書及びその中国語訳を提出しなけばならない。
2、申請者または生産企業の証明書類
2.1、申請者資格の証明書類
2.1.1、申請者としての国内機構の合法的登記証明書類(営業許可証等)、ワクチンも生産範囲に含まれるという関連医薬品生産許可証及びその変更記録ページ(上市許可申請に適用する)。![]()
2.1.2、海外申請者が中国国内の企業法人を指定して関連ワクチン登録を代理してもらう場合、委託書類、公証文書及びその中国語訳、並びに登録代理機構の営業許可証の写しを提供しなければならない。上市許可申請時に、登録代理機構を変更する場合には、海外申請者が元の委託代理関係を解除した書類、公証文書及びその中国語訳を提出しなければならない。
2.2、生産企業資格の証明書類
2.2.1、国内生産企業機構の合法的登記証明書類(営業許可証等)、ワクチンも生産範囲に含まれるという関連医薬品生産許可証及び変更記録ページ(上市許可申請に適用する)。
2.2.2、海外の医薬品管理機関の発行し、当該ワクチンの生産工場と包装工場が医薬品生産品質管理規範に適合すると証明書類、公証認証文書及びその中国語訳(海外生産医薬品に適用する)。
2.2.3、臨床試験を申請する場合には、その臨床試験用医薬品が医薬品生産品質管理規範に適合する条件の下で調製されたという状況説明書類を提供しなければならない。
2.3、小型・零細企業申請資料(適用する場合のみ)について。当該企業の営業許可証副本のコピー、 前年度企業所得税納税申請表(税務機関が確認した上で、押印したものでなければならない)または前年度有効統計表(統計機関の発行したもの)の原本。
2.4、重要な原材料、アジュバント出所、生産用毒素と生産用細胞質マトリックスの由来等を証明する書類は、3.2.S.2.3で提供しなければならない。
2.5、ワクチン臨床試験の主要担当機構は、自身が三級医療機構または省級以上の疾病予防コントロール機構であるという資格証明書類を提供しなければならない。
3、その他の申請資料![]()
3.1、登録分類及びその根拠
申請者は「生物製品登録分類及び申請資料要求」第一部の予防用生物製品の登録分類要求に準じて登録分類を明確化し、様式1の説明書類で分類の根拠を説明しなければならない。
医薬品登録分類は上市申請時に確定され、審査過程でその他の医薬品が国内外で上市したことによって変更することはない。審査承認過程で登録分類が実際の状況と合致しないと発見した場合、申請者は主動に申請を撤回し完全に修正してから改めて申請しなければならない。
3.2薬学申請資料の要求
ワクチン研究開発の法則に基づき、申請の異なる段階で、生産技術と手順、品質管理を含む薬学研究は段階的に進歩し、改善する過程である。ワクチンによってその薬学的特徴も異なる。申請者は申請資料として提出が求められたある項目の研究または一部の研究の成果を提出する必要がないと判断した場合、不適用と明記し、十分な根拠も提示しなければならない。
3.2.1、シードロット及び細胞質マトリックス
ウイルス毒素に係わるワクチン申請資料については、3.2.S.2.3部分で生産用毒素資料を提出しなければならない。3.2.S.2.3で中検院または関連医薬品監督管理機関に認められた第三者検定機関の作成した生産用菌(ウィルス)株シードロットと生産用細胞質マトリックスのシードロットに関する再審査検定報告書を提供する。
3.2.2、アジュバント
アジュバントに関する研究資料は2つの部分に分けて提出する。3.2.Pでアジュバントの概要を提出する。3.2.A.3で薬学研究に関する完全な情報を提出する。
3.2.3、外因性因子安全性評価
対象ウイルス不活化の検証資料は3.2.S.2.5生産手順検証の部分で提出する。非対象ウイルス除去または不活化の検証研究資料は3.2.A.2外因性因子安全性評価の部分で提出する。
3.2.4、多種混合/多価ワクチン
多価ワクチンについては、各種成分の生産技術と品質管理がやや類似している場合、同じ3.2.S章でまとめて記述することができる。大きく異なる場合、それぞれ単独で3.2.S章を提出する。製品に複数の成分が含まれている場合(多種混合ワクチン、または付属希釈剤)、成分ごとに原液及び(または)製剤の章を提供することができる。
3.2.5、地域情報
3.2.R.1 生産技術と手順の検証
生産技術と手順を検証する計画と報告書を提供する。
3.2.R.2 ロット単位の記録
臨床試験申請時に、臨床試験用サンプルの技術を代表するロット単位の生産、検査記録を提供する。
上市申請時に、重要臨床使用医薬品を代表するロット、及び少なくとも連続した3ロットの上市規模予定品検証ロットのロット生産と検査記録を提供する。上記ロット単位の検査報告書を提供する。
3.2.R.3 分析方法検証報告書
代表的なクロマトグラフを含む分析方法検証報告書を提供する。
3.2.R.4 安定性クロマトグラフ
安定性研究の代表的なクロマトグラフを提供する。
3.2.R.5 比較可能な実施案(該当する場合のみ)
3.2.R.6 その他
3.3、非臨床資料の要求
3.3.1、アジュバント
アジュバントについては、薬物動態学、毒性学に関する研究が行われている場合、ICH M4の基本内容に基づいて対応する部分に入れて提出する。アジュバントの類型、アジュバント使用の必要性、アジュバントまたは抗原混合比の合理性、アジュバントの作用機序などの研究内容は4.2.1.1の主要薬効学部分で提出する。
3.3.2、多種混合ワクチンまたは多価ワクチン
多種混合ワクチンまたは多価ワクチン抗原混合比の合理性、多価ワクチン抗体交差活性保持に関する研究内容は4.2.1.1の主要薬効学部分で提出する。
3.4、臨床資料の要求
3.4.1、海外の申請者が国内で未成年者向けワクチンの臨床試験を行う場合には、少なくとも海外被験者グループに関する第I相臨床試験のデータを取得しなければならない。重大且つ突発的な公共衛生事件に対応するために、緊急ニーズのあるワクチンまたは国務院衛生健康主管部署の認定した緊急必要となるワクチンは除外する。
3.4.2、臨床研究報告書は、関連ガイドラインの要求に適合しなければならない。その表紙では、医薬品登録申請者(署名及び押印)、主要または協力研究者(署名)、研究担当または協力機関の名称、統計学責任者(署名)と統計機関の名称及びICH E3で求められたその他の情報を提供しなければならない。臨床研究報告書の付録では、申請者側の医学専門家が署名しなければならない。
3.4.3、臨床試験データベースの電子ファイルについて。SAS XPORT伝送様式(即ち、xpt様式)でなければならない。ロックされたデータベースディスク1式2部を用意し、且つ別々にディスクケースに入れる。ディスクケースにはファイルタイプとしてデータベースを明記すると同時に、品名、申請者(申請者またはは登録代理機構の印鑑を押捺しなければならない)、統計ソフトウェア名称、データ管理機関、データ統計機関なども明記しなければならない。資料の表紙に品名、申請者(申請者または登録代理機構の印鑑を押捺しなければならない)を明記し、申請資料の原本とともに提出する。
3.5、使用予定の医薬品一般名称が国家医薬品基準または医薬品登録基準に収録されていない場合には、医薬品上市許可申請と同時に、一般名称認定申請も行い、一般名称認定関連資料を単独のファイル袋に入れて提出しなければならない。
3.6、医薬品医療機器コンビネーション製品の登録申請を予定しているが、同類製品の属性が医薬品と判定された場合には、医薬品として申請を行う。属性が判定されていない場合には、申請者は登録申請を行う前に、国家医薬品監督管理局に製品属性の判定を申し込まなければならない。また、属性が医薬品をメインとすると判定された場合には、「医薬品登録管理規則」に規定された手順に従って登録を行い、その医療機器の部分は医療機器登録申請資料の要求を参照し申請資料を作成し、資料を単独のファイル袋に入れて提出しなければならない。
3.7、申請者が登録申請を取り下げた後に再申請する場合、再申請の理由または補充、改善関連状況について詳細に説明しなければならない。
(五)その他の注意事項
1、海外生産ワクチンについては、海外医薬品管理機関の発行した証明書類(ワクチン上市販売許可証明書類、医薬品生産品質管理規範を適合する証明書類及び医薬品変更証明書類等を含む)を提出する場合、それらの書類は世界保健機関の推薦する統一様式を使用した原本であれば、所在国の公証機関による公証及び所在国に駐在する中国大使館・領事館の認証を受けないことができる。
2、申請者は30日以内に補正資料を完成しなければならない。申請者が正当な理由なく、期限を過ぎても補正資料を提出しなかった場合、申請を自らあきらめたと見なされ、申請資料も申請者に返却される。
五、受理審査の決定
(一)受理
1、受理通知書。形式審査の要求に適合する場合、「受理通知書」を1式2部発行する。1部は申請者に送付し、1部は資料として保存する。
2、納付通知書。費用を納付する必要な場合。
(二)補正
申請資料には不備があり、またはその形式が法定形式に合致しない場合、補正が必要な部分を一括で申請者に告知し、「補正通知書」を発行する。
(三)不受理
要求に適合しない場合、「不受理通知書」を発行し、且つ理由を説明する。
(四)受理フローチャート
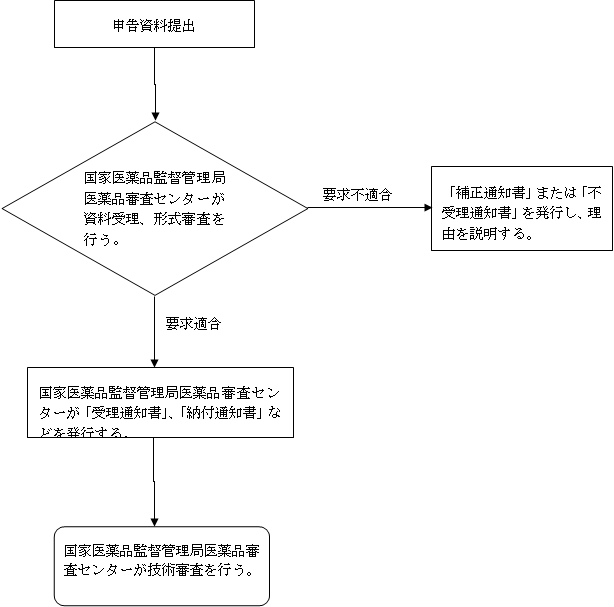
六、その他
その他の事項については、「医薬品登録管理規則」等の現行規定、技術ガイドラインを参照するように。元食品医薬品監督管理総局の2017年11月30日に発布した「医薬品登録受理と審査マニュアル(試行版)の通告」(2017年第194号)は同時に廃止する。
七、付録
1、予防用生物由来製品登録申請資料自己チェック表
別添
生物製品登録受理審査マニュアル(試行版)
第二部 治療用生物製品
本マニュアルは現行の法律、法規の要求に基づいて制定する。本マニュアルで言及されず、または明確に規定されていない受理関係事項については、申請者は受理機関と意思疎通を行うことができる。今後、本マニュアルは関連法律、法規などの書類の要求に基づいて随時更新する。
一、適用範囲
治療用生物製品の臨床試験申請または上市許可申請。
二、受理機関
国家医薬品監督管理局医薬品審査センターが受理する。
三、資料に関する基本的要求
「医薬品登録管理規則」及び「生物製品登録分類及び申請資料要求」の規定に基づき、要求に適合する申請資料を提供する。申請資料は「M4:ヒト用医薬品承認申請のための国際共通化技術資料(CTD))」(以下「CTD」と略称)の様式に基づいてまとめなければならない。また、目録及び項目番号は変更してはならず、関連情報または研究資料がない項目があっても、その番号及び名称を保留すべきで、その下に「不適用」と明記し、理由を説明する。
(一)申請表のまとめ
医薬品登録申請表、申請資料自主チェック表、小型・零細企業向け費用徴収優遇政策利用申請表(適用する場合のみ)は申請資料の部数と一致し、少なくともそのうちの一部は原本である。記入は正確、完全、規範的でなければならず、手書きまたは修正が禁止で、且つ申請表記入に関する説明の要求に適合していなければならない。
新版医薬品登録申請表作成・提出プログラムの使用に関する公告に基づき、申請表の記入とは国家医薬品監督管理局の統一的に発布したしたソフトウェアを使用し、新版「医薬品登録申請表作成・提出プログラム」で作成した電子版及び紙版の書類を提出しなければならない。使用中のバージョンが最新バージョン(最新の公告に準じる)であること、及び生成される電子ファイルがRVT様式であることを確認しなければならない。各ページのデータ照合コードは一致し、且つ提出された電子版申請表と一致していなければならない。申請表及び自主チェック表の各ページの端には、申請者または登録代理機構の割印を押さなければならない。
(二)申請資料の整理
2セットの完全な申請資料(少なくとも1セットは原本)+lセットの総括資料(様式1、様式2を含まなければならない)。各セットに関連申請表及び目次を入れる。
「医薬品登録申請表」及び検定機関の発行した検定報告書を除き、申請資料(クロマトグラフを含む)は各表紙に申請者または登録代理機構の印鑑を押捺し、且つ文字のあるところに押印しなければならない。資料整理と整理規範の詳細は「医薬品登録申請資料様式、体裁及び整理規範」を参照するように。
四、形式審査の要点
(一)申請事項審査の要点
1、臨床試験実施が承認された医薬品で、適応症(または主要治療効能)を追加し、及びその他の医薬品との併用を行う予定がある場合、申請者は新たに医薬品臨床試験申請を行わなければならない。上市が認可された医薬品で、適応症(または主要治療効能)を追加するために、医薬品臨床試験を実施する必要がまだある場合、新たに医薬品臨床試験申請を行わなければならない。
2、医薬品臨床試験終了後、医薬品臨床試験を再開して継続しようとする場合、改めて医薬品臨床試験申請を行わなければならない。医薬品臨床試験申請が承認された日から3年以内に、被験者が説明同意書に署名していない場合、当該医薬品臨床試験許可は自動的に失効する。医薬品臨床試験を実施する必要がまだある場合、改めて申請しなければならない。
3、医薬品臨床試験申請と医薬品上市許可申請は、申請した適応症に基づいて管理される。同一医薬品の異なる適応症については、それぞれ登録申請を行わなければならない(バイオシミラーは除外)。同一適応症が複数の臨床試験プロトコルの申請に係わる場合、申請表におけるその他の特別説明事項で簡単に説明しなければならない。
4、関連規定に従って臨床試験が免除された筋肉注射に用いられる一般または特異性を持つ人免疫グロブリン、人血清アルブミン等は、直接上市申請を行うことができる。
5、医薬品上市許可申請審査期間において、医薬品の安全性、有効性及び品質制御可能性に影響を及ぼす可能性のある重大変更が発生した場合には、申請者は元の登録申請を取り下げ、補足研究した後に改めて申請しなければならない。申請者の名称変更、登録住所の名称変更など技術審査内容に係わらない場合、速やかに書面で医薬品審査センターに告知し、且つ関連証明資料を提出しなければならない。
(二)意思疎通交流審査の要点
1、申請者は初回新薬臨床試験申請を行う前に、医薬品審査センターに意思疎通交流会議の開催を申し込まなければならない。
2、上市許可申請を行う前に、申請者は医薬品審査センターに意思疎通交流会議の開催を申し込まなければならない。
3、意思疎通交流は、「医薬品研究開発と技術審査関連意思疎通交流管理規則」の規定に適合しなければならない。
4、意思疎通交流を申し込んだ場合、当該申請に関連する意思疎通交流番号、意思疎通交流に関する回答意見書を提出し、意見書における質問項目に各項目ごとに回答しなければならない。
(三)申請表審査の要点
1、医薬品登録申請表
申請表は医薬品登録申請表記入説明の要求に基づいて規範的に記入し、記入した情報は証明書類における関連内容及び申請資料と一致しなければならない。
1.1、医薬品上市登録プロセスの加速について。当該申請の実際状況に基づいて選択肢にチェックを入れる。意思疎通交流を経て確認した後、「優先審査承認手順」を選択した場合には、医薬品上市許可申請を行うと同時に、「優先審査承認業務手順」に従って優先審査承認を申請する。「特別審査承認手順」を選択した場合には、「医薬品特別審査承認手順」に従って対処しなければならない。
1.2、申請事項について。当該申請の実際の申請事項に基づいて記入する。臨床研究を申請する場合、臨床試験を選択する。上市を申請する場合、上市許可を選択する。
1.3、医薬品登録分類について。「生物製品登録分類及び申請資料要求」における治療用生物製品登録分類要求に従って登録分類を明確化しなければならない。
1.4、その他の事項について。小型・零細企業の条件に合致する企業が費用徴収優遇政策利用を申請する場合、小型・零細企業費用徴収優遇を選択できる。
1.5、医薬品一般名称について。国家医薬品基準または医薬品登録基準に収録された医薬品一般名称を使用しなければならない。国家医薬品基準または医薬品登録基準に収録されていない名称を使用する場合、申請者は医薬品上市許可申請を行う際に、一般名称証明書類の原本を提出するか、同時に一般名称認定申請を行わなければならない。
1.6、同一品目の受理済または同時申請したその他の製剤及び規格について。当該品目の受理済または同時申請した製剤、あるいは規格が異なる品目の受理番号及び名称、併用する製剤の受理番号及び名称を含んで記入する。臨床研究を完了し、上市を申請する場合には、元の臨床申請受理番号、臨床試験承認番号、臨床試験登録番号を記入しなければならない。
![]()
1.7、原薬、添加剤と包装資材の出所について。医薬品登録申請を行う際には、使用する添加剤、包装資材に関する情報を全て記入しなければならず、且つ記入した情報は提出した証明書類または原薬、医薬品用添加剤と包装資材登録情報公示プラットフォームに登記した内容と一致しなければならない。
1.8、主要適応症または主要治療効能について。主要適応症または主要治療効能を簡潔に記入し、申請資料における適応症情報をすべて含まなければならない。適応症の分類は当該適応症に合致しなければならない。
1.9、今回の申請の回数について。関連品目の申請は今回で何回になるかを記入する。過去の申請及び審査承認状況を簡単に説明する。例えば、申請者が自ら撤回し、または資料が審査承認の要求に適合しないため、国家医薬品監督管理局に承認されなかった等の状況。元の申請の審査承認が終了した後、再度申請できる。
1.10、申請者及び委託研究機構
記入された情報は証明書類と申請資料と一致しなければならない。そのうちの一つの申請機構を国に登録費用を納付する業務の担当者として指定する。申請者として記入された各機構は、いずれもその法定代表者またはその授権を受けた者(署名済授権書の原本を別途提供する必要がある)が署名し、機構の公印(機構名称と完全に一致しなければならない)を押捺しなければならない。
2、小型・零細企業向け費用徴収優遇政策利用申請表
小型・零細企業向け行政事業費用徴収優遇政策の利用条件に合致する場合、小型・零細企業向け費用徴収優遇政策利用申請表を提出し、以下の情報も提供する。
2.1、基本情報。例えば企業名称、連絡担当者、連絡用電話番号等。いずれも「医薬品登録申請表」の関連情報と一致しなければならない。![]()
2.2、従業員、前納税年度の営業収入、企業資産総額等。申請者は実際の状況に基づいて記入する。
2.3、その法定代表者またはその授権を受けた者(署名授権書の原本を別途提供する必要がある)が署名し、機構の公印(機構名称と完全に一致しなければならない)を押捺しなければならない。
(四)申請資料審査の要点
国家医薬品監管機関の発布した証明事項の取消しに関する公告に「内部確認審査へ変更」と規定された証明事項については、公告の要求に準じる。
1、製品関連証明書類
1.1、医薬品用添加剤及び包装資材の証明書類。
1.1.1、医薬品用添加剤及び包装資材の合法的な出所に関する証明書類。供給契約書、領収書等が含まれる(登録済原薬、医薬品用添加剤及び包装資材を選択して使用しない製剤の場合に適用する)。
1.1.2、医薬品用添加剤及び包装資材使用授権書の写し(製剤が登記済原薬、医薬品用添加剤及び包装資材を選択して使用するの場合に適用する)。サプライヤーが当該書類を提供する場合、医薬品用添加剤と包装資材生産企業の授権が必要で、授権書類のコピーを添付しなければならない。
1.2、特許情報及び証明書類
申請対象となる医薬品または使用する処方箋、生産技術と手順、用途などに関する特許状況及びその権利帰属状況の説明文、並びに他人の特許権を侵害しない旨の誓約書。申請者が作成し、権利侵害の可能性がある場合には、全責任を負うと約束しなければならない。
1.3、対照薬出所を証明する書類(該当する場合のみ)。
1.4、商標情報及び証明書類(該当する場合のみ)。
商品名の使用を申請する場合、商標登録証の写しを提供しなければならない。
1.5、医薬品臨床試験に関する証明書類(上市許可申請に適用する)
「医薬品臨床試験承認書」または臨床試験通知書及び臨床試験実施プロセスにおける関連補充申請(あれば、提出する)書類のコピー、臨床試験用医薬品品質基準及び臨床試験登録番号等の関連資料並びに「医薬品臨床試験承認書」または臨床試験通知書における意見について各項目ごとに回答しなければならない。
1.6、研究機構資格の証明書類
非臨床研究安全性評価機構は、医薬品監督管理機関の発行した「医薬品非臨床研究品質管理規範」(略称はGLP)に適合する承認証明または査察報告書等の証明書類を提供しなければならない。臨床試験機構は届出証明書類を提供しなければならない。
1.7、医薬品・医療機器コンビネーション製品に関する証明書類
属性判定により、医薬品に属し、または医薬品をメインとする医薬品・医療機械コンビネーション製品であると確認された場合、医薬品・医療機器コンビネーション製品属性判定結果通知書の写しを提出しなければならない。
1.8、医薬品上市販売許可証明書類。
海外上市済の生物製品は、海外医薬品管理機関の発行した医薬品上市販売許可書類、公証認証文書及びその中国語訳を提出しなけばならない。
2、申請者または生産企業資格の証明書類
2.1、申請者資格の証明書類
2.1.1、申請者としての国内機構の合法的登記証明書類(営業許可証等)、申請対象となる医薬品も生産範囲に含まれるという生産許可証及びその変更記録ページ(上市許可申請に適用する)。
2.1.2、海外申請者が中国国内の企業法人を指定して関連医薬品登録を代理してもらう場合、委託書類、公証文書及びその中国語訳、並びに登録代理機構の営業許可証の写しを提供しなければならない。上市許可申請時に、登録代理機構を変更する場合には、海外申請者が元の委託代理関係を解除した書類、公証文書及びその中国語訳を提出しなければならない。
2.2、生産企業資格の証明書類
2.2.1、国内生産企業と機構の合法的登記証明書類(営業許可証等)、申請対象となる医薬品も生産範囲に含まれるという生産許可証及び変更記録ページ(上市許可申請に適用する)。
2.2.2、海外の医薬品管理機関の発行した当該医薬品の生産工場と包装工場が医薬品生産品質管理規範に適合すると証明する書類、公証認証文書及びその中国語訳(海外生産医薬品に適用する)。
2.2.3、臨床試験を申請する場合には、その臨床試験用医薬品が医薬品生産品質管理規範に適合する条件の下で調製されたという状況説明書類を提供しなければならない。
2.3、小型・零細企業申請資料(適用する場合のみ)について。当該企業の営業許可証副本のコピー、 前年度企業所得税納税申告表(税務機関が確認した上で、押印したものでなければならない)または前年度有効統計表(統計機関の発行したもの)の原本。
3、その他の申請資料審査の要点
3.1、登録分類及びその根拠
申請者は「生物製品登録分類及び申請資料要項」第二部の治療用生物製品の登録分類要求に準じて登録分類を明確化し、様式1の説明書類で分類の根拠を説明しなければならない。
医薬品登録分類は上市申請時に確定され、審査過程でその他の医薬品が国内外で上市したことによって変更することはない。審査承認過程で登録分類が実際の状況と合致しないと発見した場合、申請者は主動に申請を撤回し、完全に修正した後に改めて申請しなければならない。
3.2、薬学申請資料の要求
医薬品研究開発の規則に基づき、申請の異なる段階で、生産技術と手順、品質管理を含む薬学研究は段階的に進歩し、改善する過程である。生物製品によってその薬学的特徴も異なる。申請者は申請資料として提出が求められたある項目の研究または一部の研究の成果を提出する必要がないと判断した場合、該当しないと明記し、十分なサポート根拠も提示しなければならない。
3.2.1、バイオシミラーについては、「3.2.R.6その他の書類」で品質類似性評価の資料を提出する。
3.2.2、抗体薬物複合体または修飾剤については、小分子薬物の薬学研究資料はCTD様式と内容に関する要求に準じて単独で1セットの資料を提出し、もしくは「3.2.S.2.3物質材料管理」ですべての薬学研究資料を提出することができる。
3.2.3、複方製品または複数成分含有製品については、成分ごとに原液及び(または)製剤の章を提供することができる。
3.2.4、細胞と遺伝子治療製品については、製品の特徴によって、原液及び(または)製剤に関する部分で薬学研究資料を提出できる。適用しない場合、「不適用」と明記するように。
3.2.5、地域情報
3.2.R.1 生産技術と手順の検証
生産技術と手順を検証する計画書と報告書を提供する。
3.2.R.2 ロット単位の記録
臨床試験申請時に、臨床試験用サンプルの技術を代表するロット単位の生産、検査記録を提供する。上市申請時に、重要臨床使用医薬品を代表するロット、及び少なくとも連続した3ロットの上市規模検証ロット予定品の生産と検査記録を提供する。上記ロット単位の検査報告書を提供する。
3.2.R.3 分析方法検証報告書
代表的なクロマトグラフを含む分析方法検証報告書を提供する。
3.2.R.4 安定性クロマトグラフ
安定性研究の代表的なクロマトグラフを提供する。
3.2.R.5 比較可能な実施案(該当する場合のみ)
3.2.R.6 その他
3.3、臨床資料の要求
3.3.1、臨床研究報告書は、関連ガイドラインの要求に適合しなければならない。その表紙では、医薬品登録申請者(署名及び押印)、主要または協力研究者(署名)、研究担当または協力機関の名称、統計学責任者(署名)と統計機関の名称及びICH E3で求められたその他の情報を提供しなければならない。臨床研究報告書の付録Ⅱでは、申請者側の医学専門家が署名しなければならない。
3.3.2、臨床試験データベースの電子ファイルについて。SAS XPORT伝送様式(即ち、xpt様式)でなければならない。ロックされたデータベースディスク1式2部を用意し、且つ別々にディスクケースに入れる。ディスクケースにはファイルタイプとしてデータベースを明記すると同時に、品名、申請者(申請者またはは登録代理機構の印鑑を押捺しなければならない)、統計ソフトウェア名称、データ管理機関、データ統計機関なども明記しなければならない。ファイル袋の表紙に品名、申請者(申請者または登録代理機構の印鑑を押捺しなければならない)を明記し、申請資料の原本とともに提出する。
3.4、使用予定の医薬品一般名称が国家医薬品基準または医薬品登録基準に収録されていない場合には、医薬品上市許可申請と同時に、一般名称認定申請も行い、一般名称認定関連資料を単独のファイル袋に入れて提出しなければならない。
3.5、医薬品・医療機器コンビネーション製品の登録申請が予定しているが、同類製品の属性が医薬品と判定された場合には、医薬品として申請を行う。属性が判定されていない場合には、申請者は登録申請を行う前に、国家医薬品監督管理局に製品属性の判定を申し込まなければならない。また、属性が医薬品をメインとすると判定された場合には、「医薬品登録管理規則」に規定された手順に従って登録を行い、その医療機器の部分は医療機器登録申請資料の要求を参照し申請資料を作成し、資料を単独のファイル袋に入れて提出しなければならない。
3.6、申請者が登録申請を取り下げた後に再申請する場合、再申請の理由または補充、改善関連状況について詳細に説明しなければならない。
(五)その他の注意事項
1、生物製品に属する体内診断用試薬は「M4:ヒト用医薬品承認申請のための国際共通化技術資料(CTD))」を参照して申請資料を作成する。
2、臨床試験の実施または上市が許可された生物製品で、適応症追加のための臨床試験申請を行う場合、初申請と重複する資料(様式1を除く)の提出が免除される。但し、初申請関連資料の番号を今回の申請資料に記載しなければならない。
3、血漿採集ステーションは国家衛生行政管理機関の発布した「血漿採集ステーション品質管理規範」の要求に適合しなければならない(関係のある場合のみ)。 2、臨床試験の実施または上市が許可された生物製品で、適応症追加のための臨床試験申請を行う場合、初申請と重複する資料(様式1を除く)の提出が免除される。但し、初申請関連資料の番号を今回の申請資料に記載しなければならない。
4、海外生産医薬品については、海外医薬品管理機関の発行した証明書類(医薬品上市販売許可証明書類、医薬品生産品質管理規範への適合を証明する書類及び医薬品変更許可証明書類等を含む)を提出する場合、それらの書類は世界保健機関が推薦する統一様式を使用した原本であれば、所在国の公証機関による公証及び所在国に駐在する中国大使館・領事館の認証を受けないことができる。
5、申請者は30日以内に補正資料を完成しなければならない。申請者が正当な理由なく、期限を過ぎても補正資料を提出しなかった場合、申請を自らあきらめたと見なされ、申請資料も申請者に返却される。
五、受理審査の決定
(一)受理
1、受理通知書。形式審査の要求に適合する場合、「受理通知書」を1式2部発行する。1部は申請者に送付し、1部は資料として保存する。
2、納付通知書。費用納付必要な場合。
(二)補正
申請資料には不備があり、またはその形式が法定形式に合致しない場合、補正が必要な部分を一括で申請者に告知し、「補正通知書」を発行する。
(三)不受理
要求に適合しない場合、「不受理通知書」を発行し、且つ理由を説明する。
(四)受理フローチャート
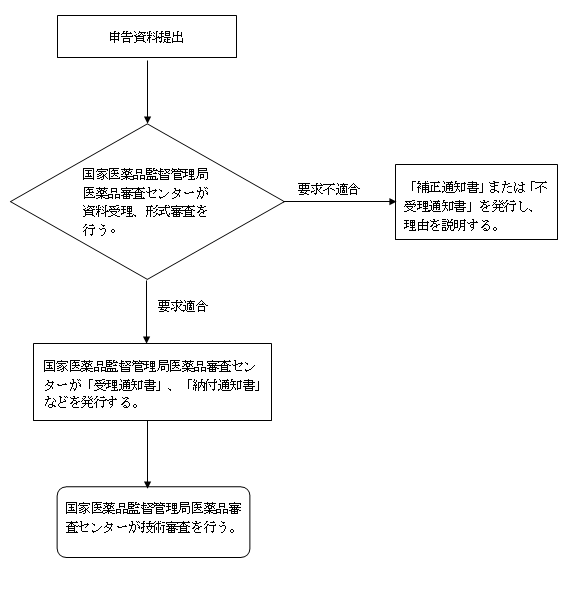
六、その他
その他の事項については、「医薬品登録管理規則」等の現行規定、技術ガイドラインを参照するように。元食品医薬品監督管理総局の2017年11月30日に発表した「医薬品登録受理審査マニュアル(試行版)の発布に関する通告」(2019年第194号)は同時に廃止する。
七、付録
1、治療用生物由来製品登録申請資料自己チェック表
2、参考目録
別添
生物製品登録受理審査マニュアル(試行版)
第三部 生物製品として管理される体外診断用試薬
本マニュアルは現行の法律、法規の要求に基づいて制定する。本マニュアルで言及されず、または明確に規定されていない受理関係事項については、申請者は受理機関と意思疎通を行うことができる。今後、本マニュアルは関連法律、法規などの書類の要求に基づいて随時更新する。
一、適用範囲
生物製品として管理される体外診断用試薬の上市許可申請。
注:生物製品として管理される体内診断用試薬は治療用生物製品受理審査マニュアルの要求を参照するように。
二、受理機関
国家医薬品監督管理局医薬品審査センターが受理する。
三、申請資料に関する基本的要求
「医薬品登録管理規則」及び「生物製品登録分類及び申請資料要求」の規定に基づき、要求に適合する申請資料を提供する。申請資料の様式、目次及び項目番号は変更してはならず、関連情報または研究資料がない項目があっても、その番号及び名称を保留すべきで、その下に「不適用」と明記し、理由を説明する。
(一)申請表の整理
医薬品登録申請表、申請資料自主チェック表、は申請資料の部数と一致し、少なくともそのうちの一部は原本である。記入は正確、完全、規範的でなければならず、手書きまたは修正が禁止で、且つ申請表記入に関する説明の要求に適合していなければならない。
新版医薬品登録申請表作成・提出プログラムの使用に関する公告に基づき、申請表の記入と提出は国家医薬品監督管理局の統一的公表したフトウェアを使用し、新版「医薬品登録申請表作成・提出プログラム」で作成した電子版及び紙版の書類を提出しなければならない。使用中のバージョンが最新バージョン(最新の公告に準じる)であること及び生成される電子ファイルがRVT様式であることを確認しなければならない。各ページのデータ照合コードは一致し、且つ提出された電子版申請表と一致していなければならない。申請表及び自主チェック表の各ページの端には、申請者または登録代理機構の割印を押さなければならない。
(二)申請資料の整理
2セットの完全な申請資料(少なくとも1セットは原本)+lセットの総括資料(。各セットに関連申請表及び目次を入れる。
「医薬品登録申請表」及び検定機関の発行した検定報告書を除き、申請資料(クロマトグラフを含む)は各表紙に申請者または登録代理機構の印鑑を押捺し、且つ文字のあるところに押印しなければならない。資料整理と装丁要求の詳細は「医薬品登録申請資料様式、体裁と整理規範」を参照するように。
四、形式審査の要点
(一)申請事項審査の要点
生物製品として管理される体外診断用試薬については、申請者は国内で臨床試験を完了した後、医薬品上市許可申請を直接行うことができる。
(二)意思疎通交流審査の要点
意思疎通交流を申し込んだ場合、申請時に発行された意思疎通交流番号、意思疎通交流に関する回答意見書を提出し、意見書における質問項目に各項目ごとに回答しなければならない。
(三)申請表審査の要点
申請表は医薬品登録申請表記入説明の要求に基づいて規範的に記入し記入した情報は証明書類と申請資料と一致しなければならない。
1、医薬品上市登録プロセスの加速について。当該申請の実際状況に基づいて選択肢にチェックを入れる。意思疎通交流をへて確認した後、「優先審査承認手順」を選択した場合には、医薬品上市許可申請を行うと同時に、「優先審査承認業務手順」に従って優先審査承認を申請する。「特別審査承認手順」を選択した場合には、「医薬品特別審査承認手順」に従って対処しなければならない。
2、申請事項について。上市を申請する場合、上市許可を選択する。
3、医薬品登録分類について。「生物製品登録分類及び申請資料要求」における生物製品として管理される体外診断用試薬登録分類の要求に準じて登録分類を明確化する。
4、医薬品一般名称について。国家医薬品基準または医薬品登録基準に収録された医薬品一般名称を使用しなければならない。国家医薬品基準または医薬品登録基準に収録されていない名称を使用する場合、申請者は医薬品上市許可申請を行う際に、一般名称証明書類の原本を提出するか、同時に一般名称認定申請を行わなければならない。
5、今回の申請の回数について。関連品目の申請は今回で何回目になるかを記入する。過去の申請及び審査承認状況を簡単に説明する。例えば、申請者が自ら撤回し、または資料が審査承認の要求に適合しないため、国家医薬品監督管理局に承認されなかった等の状況。元の申請の審査承認が終了した後、再度申請できる。
(四)申請資料審査の要点
国家医薬品監管機関の公表した証明事項の取消しに関する公告に「内部確認審査へ変更」と規定された証明事項については、公告の要求に準じる。
1、製品関連証明書類
1.1、特許情報及び証明書類
申請対象となる医薬品または使用する処方箋、生産技術と手順、用途などに関する特許状況及びその権利帰属状況の説明文、並びに他人の特許権を侵害しない旨の誓約書。申請者が作成し、権利侵害の可能性がある場合には、全責任を負うと約束しなければならない。
1.2、商標情報及び証明書類。
商品名の使用を申請する場合、商標登録証を提供しなければならない。
1.3、上市販売許可証明書類
海外上市済体外診断用試薬の場合、海外医薬品監督管理機関の発行した当該試薬上市販売許可証明書類、公証認証文書及びその中国語訳を提出しなければならない。
2、申請者または生産企業の証明書類
2.1、国内申請者と生産企業または機構の合法的登記証明書類(営業許可証等)、申請対象となる医薬品の生産許可証及び変更記録ページ。
2.2、海外生産診断用試薬の生産工場と包装工場が医薬品生産品質管理規範に適合すると証明する書類、公証認証文書及びその中国語訳。海外申請者が中国国内の企業法人を指定して関連医薬品登録を代理してもらう場合、委託書類、公証文書及びその中国語訳、並びに登録代理機構の営業許可証の写しを提供しなければならない。
3、その他の申請資料審査の要点![]()
3.1、登録分類及びその根拠
申請者は「生物製品登録分類及び申請資料要求」第三部の生物製品として管理される体外診断用試薬の登録分類要求に準じて登録分類を明確化し、研究目的と根拠の部分で分類の根拠を説明しなければならない。
医薬品登録分類は上市申請時に確定され、審査過程でその他の医薬品が国内外で上市したことによって変更することはない。審査承認過程で登録分類が実際の状況と合致しないと発見した場合、申請者は主動に申請を撤回し、完全に修正した後に改めて申請しなければならない。
3.2、臨床研究報告書は、関連ガイドラインの要求に適合しなければならない。その表紙では、医薬品登録申請者(署名及び押印)、主要または協力研究者(署名)、研究担当または協力機関の名称、統計学担当者(署名)と統計機関の名称及びICH E3で求められたその他の情報を提供しなければならない。臨床研究報告書の付録では、申請者側の医学専門家が署名しなければならない。
3.3、使用予定の医薬品一般名称が国家医薬品基準または医薬品登録基準に収録されていない場合には、医薬品上市許可申請と同時に、一般名称認定申請も行い、一般名称認定関連資料を単独のファイル袋に入れて提出しなければならない。
3.4、申請者が登録申請を取り下げた後に再申請する場合、再申請の理由または補充、改善関連状況について詳細に説明しなければならない。
(五)その他の注意事項
1、少なくとも3社の国内臨床試験機構で臨床試験を完了し、臨床試験契約書及び臨床試験プロトコルを提供する。海外申請者は海外で完了した臨床試験の資料、海外臨床使用状況の総括報告書と国内で完了した臨床試験の資料を提供しなければならない。
2、申請者が提供し、海外医薬品管理機関の発行した証明書類は世界保健機関の推薦する統一様式を使用しなければならない。その他の様式の書類は所在国の公証機関による公証及び所在国に駐在する中国大使館・領事館の認証を受けなければならない。
3、申請者は30日以内に補正資料を完成しなければならない。申請者が正当な理由なく、期限を過ぎても補正資料を提出しなかった場合、申請を自らあきらめたと見なされ、申請資料も申請者に返却される。
五、受理審査の決定
(一)受理
1、受理通知書。形式審査の要求に適合する場合、「受理通知書」を1式2部発行する。1部は申請者に送付し、1部は資料として保存する。
2、納付通知書。費用納付必要時。
(二)補正
申請資料には不備があり、またはその形式が法定形式に合致しない場合、補正が必要な部分を一括で申請者に告知し、「補正通知書」を発行する。
(三)不受理
要求に適合しない場合、「不受理通知書」を発行し、且つ理由を説明する。
(四)受理フローチャート
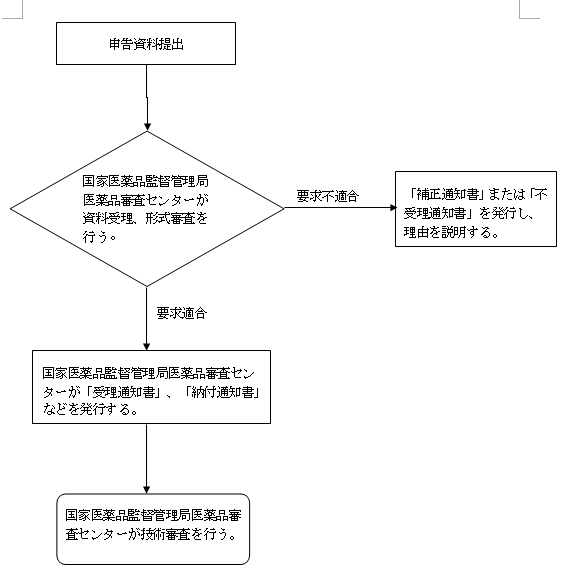
六、その他
その他の事項については、「医薬品登録管理規則」等の現行規定、技術ガイドラインを参照するように。
七、付録
1、生物由来製品として管理される体外診断用試薬登録申請資料自己チェック表
2、参考目録
(出所:医薬品審査センターウェブサイト 2020-07-03)