

 ホーム
ホーム 最新情報
最新情報 機構紹介
機構紹介 国際交流
国際交流 サイトについて
サイトについて腎不全患者における薬物動態研究技術ガイドライン
一、前書き
薬物は様々なメカニズムで体内から排除される。多くの薬物は、小腸の代謝と輸送、肝臓の代謝と輸送、および腎クリアランスを含む、以上の経路の1つまたは複数によって組み合わせて排除される。腎不全では、糸球体濾過、能動的尿細管分泌、および受動的再吸収がすべて影響を受け、腎クリアランスが低下する可能性があり、腎クリアランスの程度は通常、腎疾患の重症度によって影響を受ける。薬物が主に腎排泄によって排除される場合、腎不全(Impaired Renal Function)はその薬物動態(Pharmacokinetics、PK)に一定の変化をもたらす可能性があり、臨床投与の安全性と有効性にも影響を与える可能性がある。従って、腎不全の患者は、正常な腎機能の患者と比較して用法用量を調整する必要がある。
腎不全患者における薬物のPK変化は、主に薬物および/または代謝物の腎排泄の減少または遅延によって引き起こされる。さらに、薬物吸収、血漿タンパク質結合、膜貫通輸送、および組織分布の変化にも関連している。研究によると、腎不全は、肝臓や腸内の特定の薬物の代謝および輸送経路を変化させる可能性がある。これらの変化は、腎機能が著しく低下している患者で特に顕著であり、薬物クリアランスの主な経路が腎排泄ではない場合でも観察される可能性がある。
上記の考慮事項に基づいて、腎不全の患者に使用される可能性のあるほとんどの薬物について、合理的な用法用量の意見を提供するために、腎不全の患者のPK特性を評価することが推奨される。推奨される用法用量は、一般に、腎不全における薬物曝露の変化の程度と、曝露-反応関係(有効性と安全性を含む)に基づいて設定される。
腎不全患者を対象としたPK研究の結果は、革新的な医薬品の市販をサポートするための主要な臨床研究のための試験集団を選択するための基礎を提供することができる。腎不全患者のPK研究、曝露-反応関係研究、および市販をサポートする主要な臨床研究の被験者集団は、革新的な医薬品の市販の申請における適応集団および承認された添付文書に対応する集団、用法用量を含めることができるかどうかに影響する。
このガイドラインは、対応する集団の用法用量を調整する必要があるかどうかに関する研究データを提供するために、革新的な医薬品の開発中に腎不全の患者のPK研究に関する提案と考慮事項を提供することを目的としている。
二、基本的な考慮事項
(一)腎機能の測定
腎機能を評価する様々な方法がある。糸球体濾過量(GFR)は通常、外因性マーカー(イヌリン、イソニコチンアミド、EDTA、ジエチレントリアミン五酢酸、イオヘキソールなど)を使用して決定される。これは、式の推定よりも正確である。腎機能は、特定の時間の尿サンプルで測定されたクレアチニンクリアランス(CLCr)を使用して評価することもできる。
ただし、上記の方法は、臨床現場では日常的に使用されていない。血清クレアチニンと血清シスタチンCに基づいて確立された式を使用して腎機能を推定することがより実用的である。現在の臨床実践では、公式で推定されたGFR(eGFR)は、成人でCLCrをCockcroft-Gault(C-G)の公式で推定することを含む広範に適用されている。現在、eGFRを推定するために一般的に使用されている方法には、慢性腎臓病疫学共同研究チーム(CKD-EPI)の公式と腎臓病の食事改良試験(MDRD)の公式が含まれる。
臨床実践では、eGFRは体表面積(BSA)値1.73 m2で補正され、mL/min/1.73 m2の単位で表される。腎臓疾患の診断や治療における臨床的な効果評価、腎不全の程度の分類は、通常、BSA補正のeGFR(単位:mL/min/1.73 m2)に基づいている。ただし、腎クリアランスは絶対GFRと相関している。つまり、薬物の腎クリアランスは、BSA補正GFRではなく、個々のGFR(mL/minで表される)に比例する。したがって、用量は標準化された1.73m2のeGFRではなく、絶対GFR(mL/minで表される)に基づくことが推奨されている。個別の投与が必要な場合、補正されたeGFRに患者のBSAを掛け、1.73で割る。
腎不全患者のPK研究では、外因性マーカーを使用してGFRを正確に測定でき、式を使用してGFRを推定する方法も、被験者の腎機能を評価するために検討できる。さまざまな方法の長所と短所を考慮して、臨床的に広く受け入れられている他の方法も、評価後に適用を検討することができる。研究目的と臨床ニーズに基づいて、腎機能を測定するための合理的な方法を選択することが推奨されている。
小児患者では、腎機能も測定または推定できる。2歳未満の小児患者では、成熟度、在胎週数、およびその他の要因の影響を考慮する必要がある。Schwartz式は現在、小児GFRの推定に使用されているが、この式は新生児GFRを過大評価していると考えられており、研究者はその適用性に注意を払う必要がある。

このガイドラインの腎機能計算式は、研究者の参考のためのものであり、科学技術の継続的な発展により、臨床実践で認められている方法で腎機能を測定することができ、科学的考察とそれに対応する根拠が提供される。
(二)腎不全研究を実施するかどうかの検討
このガイドラインの独立したPK研究は、このガイドラインで説明されている完全なPK研究と省略されたPK研究を含む、集中的な採血を伴う適切に設計されたPK研究である。
治験薬および/または活性代謝物が主に腎臓から排泄される場合(例えば、尿中排泄率(fe)≥0.3)、異なる分類の腎不全の患者で独立したPK研究を実施し、対応する研究結果は異なる分類の腎不全の患者での用法用量をサポートする。一般に、分子量が69 kDa未満の治療性タンパク質及びペプチド薬は腎不全患者では腎クリアランスが低下するため、腎不全患者では分子量が69 kDa未満の治療性タンパク質やペプチド薬のPK研究を検討することが推奨されている。
主に非腎経路で除去される薬では、腎不全がPKに及ぼす影響を検討することもしばしば重要である。この場合、腎不全患者に使用される可能性のある上記の非腎排泄型薬剤について、簡略化されたPK研究を検討することができる。
以下を含むがこれらに限定されない以下の条件が満たされる場合、独立したPK研究は一般に必要とされない。
1. 主に肺から除去されるガス状または揮発性の薬物および/または活性代謝物。
2. 単回/時折の投薬のみを必要とし、薬および/または主な活性代謝物のクリアランス時間延長が安全性リスクをもたらさない薬物。
3. 分子量が69 kDaを超える治療性タンパク質薬。でも、抗体薬物複合体又は小分子に結合した他の高分子タンパク質には特別な配慮が必要である。
4. 全身吸収が制限された局所作用薬(例えば、局所薬)。
5. 主に肝臓によって除去され、安全性データから、薬物曝露が大幅に増加しても用法用量の調整が不要であることを示唆している薬物。
6. 主に肝臓によって除去され、腎不全のために薬物曝露が増加する可能性は、臨床診療で管理することができる薬物。例えば、一部の薬剤は、有効性および/または安全性に関連するマーカーを監視することによって用量滴定を行うことができる。
上記の状況について、母集団薬物動態などの他の方法を用いて研究を実施する必要があるかどうかは、ケースバイケースで分析される。
末期腎疾患(ESRD)で透析を必要とする患者に薬が投与される可能性がある場合、透析が薬およびその可能性のある活性代謝物のクリアランスに与える影響を確認するために、透析と非透析の両方でPK研究を実施する必要がある。透析療法は、薬物および/または活性代謝物の大部分を除去する可能性があり、臨床曝露量に大きな変化をもたらす。この状況では、透析後の補充などの用法用量の調整が必要である。透析患者で研究を行うときは、異なる透析療法の違いに注意を払う必要がある。透析療法が薬物またはその活性代謝物のクリアランスに有意差をもたらす可能性が低い場合、例えば、治験薬が以下の特徴を持つ場合:治験薬が透析によってクリアランスできない分子量を有する、治験薬が腎不全の影響を受けない高血漿タンパク結合率有する、以上の状況では、透析療法なしの独立したPK研究を検討することができる。
(三)試験タイミングに関する考慮事項
標的適応症集団を含む重要な臨床研究では、特定の用法用量における薬の安全性と有効性を調べることができる。通常、重度の腎不全患者は、重要な臨床研究の被験者集団から除外される。
薬の標的適応症集団に腎不全患者が比較的多く含まれ、患者が特定の治療方法の提案を必要とする場合、腎不全患者の有効性および安全性に関するデータを取得することは、高い臨床的価値を有する可能性がある。これらのデータは、腎不全患者の曝露-反応関係が変化する可能性があると予想される場合、さらに重要である。腎不全の患者が重要な臨床研究に含まれるように、前向き研究を通じて適切な用量調整を行うことを目的として、初期の薬物開発中に腎不全が薬物PKに及ぼす影響を評価することが推奨される。初期の予備評価は、第I相および/または第II相の研究から得られたデータに基づいて、又はモデリングおよびシミュレーション戦略(例えば、生理学に基づく薬物動態モデル)を用いて研究を実施することができる。これらの研究は、後続の腎不全患者PK研究の設計および重要な臨床研究に腎不全患者を組み込むことに役立つ。腎不全患者への推奨用法用量の臨床使用に関するデータと経験を取得することを容易にする。
独立したPK研究を行う必要はないが、母集団薬物動態の手法が必要である可能性を排除しない場合には、状況に応じて総合的に考慮する。
三、試験設計
腎不全患者を対象としたPK研究は、腎不全が薬物のPKに与える影響を評価し、曝露-反応関係に基づいて用法用量調整の必要性を評価するように設計される。研究は、薬物の特性や標的適応症集団の特性などの情報をもとに、さまざまな方法で実行できる。
(一)完全なPK研究の設計
1. 研究集団
治験薬PKの特性に対する腎不全の影響を十分に研究するために、通常に正常な腎機能と異なる分類の腎不全(GFR評価に基づく)の被験者を組み入れるべきである。そのうち、腎不全被験者は、慢性腎臓病(Chronic Kidney Disease、CKD。腎障害の指標があるか、GFRが60 mL/min未満で3ヶ月以上継続)および/または安定した腎機能を持つ個人である必要がある。腎不全の被験者は、血行動態が安定していることを保持する必要がある。
臨床実践で一般的に使用される糸球体濾過量の分類は次の表2に示される。
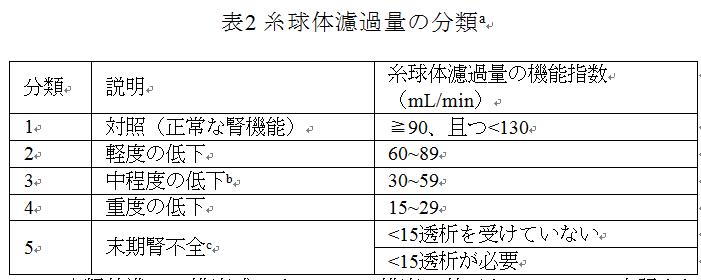
a:eGFR:分類基準は、推定式によるGFRの推定に基づき、mL/minで表記される。mL/min/1.73m2をmL/minに変換し、適切な式を用いて計算された個人BSAを掛けてから、1.73で割る。公式は次の通り:
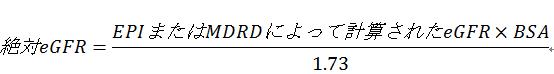
b:実際の臨床状況と研究の具体的なニーズに応じて、中等度の腎不全患者をeGFRに従って2つのグループ(45~59 mL/minと30~44 mL/min)に分けることが考えられる場合がある。
c:腎不全:この分類は専門的な腎機能障害の研究を行うためのもので、腎臓疾患の分類には適用されない。
治療ウィンドウが広い薬物のPKに対する腎不全の影響を評価するために、60 mL/min以上(正常または軽度の腎不全)、15~59 mL/min(中等度から重度の腎不全)、15 mL/min未満、または非透析日に透析を必要とする患者の分類に基づいて、被験者の層別化を考慮することができる。
薬物のPKに影響を与える可能性のある他の要因は、腎不全群と対照群で一致する必要があり、同時に、薬物のPKに影響を与える可能性のある併用薬を服用している被験者を含めるべきではない。
標的適応症集団自身が薬物のPK特性に影響を与えるいくつかの要因を持っている可能性があるため、理想的には、対照群が標的適応症の代表的なグループである。この場合、対照群は表2で定義した正常な腎機能を持つ患者でなくてもよい。
2. サンプルサイズ
各腎不全分類群の被験者数は、信頼できるPKパラメータを得るのに十分である必要があり、サンプルサイズの推定方法と結果を提出し、サンプルサイズ推定時の主要なパラメータ設定の根拠を提供する。サンプルサイズの推定では、PKパラメータの変異度が健康な人と患者の間で異なる可能性があることに注意を支払う。
3. 投与方法
標的適応症集団での研究結果より、薬物および/または主な活性代謝物が線形および非時間依存性のPK特性を示す場合、単回投与研究を検討することができる。非線形または時間依存性のPK特性を示す場合、複数回投与研究が必要である。前期の研究で推定された腎不全患者の薬物および/または主要な活性代謝物のPK特性に基づいて、最適な用法用量を評価し、選択することができる。
一般に、単回投与研究では、薬物の最高血中濃度が腎機能の影響を受けにくいことを考慮して、異なる分類の腎不全の被験者に用量を投与するか、または前期の研究データに基づいて異なる用量を投与することができる。用量選択の根拠を提供する。
複数回投与研究では、通常、定常状態に達するのに十分な長い投与期間が必要である。特に消失半減期が大幅に長い場合は、定常状態までの時間を短縮するために、負荷線量の戦略を検討することができる。複数回投与研究では、薬物とその代謝物の蓄積を減少させるために、より低い投与量またはより少ない投与頻度の研究設計を考慮できる。
4. サンプル採取及び検出分析
血液(例えば、血漿、血清または全血)サンプル、尿サンプル中の未変化体および/または調査する主要な代謝物について検査と分析を行う必要がある。血液サンプルと尿サンプルの採取頻度と時間は、未変化体および/または主要代謝物の関連PKパラメータを正確に評価できるように合理的に設計されるべきである。
腎不全患者では、薬物の血漿タンパク結合率に変化が生じることが多い。全身作用する薬物とその活性代謝物の場合、作用部位への薬物輸送の速度と程度は、血中の遊離薬物濃度に依存する。薬物のタンパク結合率が濃度に依存し、および/または代謝物やその他の経時変化要因の影響を受ける場合、薬物の総濃度を測定すると同時に、できるだけ血液サンプルごとに游離薬物濃度を測定することを勧めする。その他の場合には、限られた数又は1つの血漿サンプルを各患者から採取して、遊離薬物濃度を測定することができる。血漿タンパク結合が比較的低い薬物とその代謝物(結合が80%未満)の場合、腎不全が薬物の血漿タンパク結合率に比較的小さい影響を与え、この場合、薬物の血中総濃度を使用してそのPK特性を説明することを考慮することができる。
(二)PK研究を簡略化するための考慮事項
主に非腎経路によって除去されるが、腎不全患者に用いられる可能性のある治験薬については、簡略化されたPK研究設計を用いて、用法用量の調整が必要かどうかを検討することができる。
簡略化されたPK研究では、通常、重度の腎不全(eGFR:15~29 mL/min)の場合の薬物PKへの影響を最初に調べる。簡略化されたPK研究の結果が、重度の腎不全が薬物のPK特性を著しく変化させないことを示している場合は、さらに独立したPK研究を検討できる。簡略化されたPK研究で、重度な腎不全の被験者における薬物のPK特性の変化が臨床的な関連影響があることを示している場合、他の腎不全分類患者の臨床リスクを排除することはできず、薬物PKに対する他の腎機能分類レベル(例えば、中等度の腎不全、表2を参照)の影響をさらに評価する必要がある。このような状況では、完全なPK研究を実施することが可能であり、また、第II相または第III相臨床試験における母集団薬物動態研究のような追加の研究を実施することが可能である。
(三)第II相または第III相研究で薬物PKに対する腎不全の影響の評価
第II相および/または第III相臨床研究のデータを用いて母集団薬物動態解析を行う場合には、十分なサンプルサイズ、異なる分類の腎不全患者が必要であり、腎不全の薬物PKへの影響を説明するのに十分なデータがあり、この場合、腎機能指標を曝露-反応関係の独立した影響要因として、腎機能の薬物曝露への影響を調べる必要がある。
サンプルサイズが不十分な場合、例えば、重度の腎不全患者のサンプルサイズが不十分な場合、臨床研究に含まれていない腎不全集団に適切な用法用量を提供するために、簡略化されたPK研究が必要になる場合がある。
母集団薬物動態分析や曝露-反応分析などのモデリング分析手法を適用する場合、分析データセットに含まれる臨床研究には、このガイドラインの完全なPK研究の設計に記載されている重要な内容が含まれている必要がある。次の内容に注意を支払うことが推奨される。
1. 異なる分類の腎不全被験者の十分なサンプルサイズを設定する。
2. 薬の投与とサンプルの採取時間を正確に記録する。
3. 各被験者から十分な量のサンプルを採取する。
4. 適切な状況で遊離薬物濃度を測定する。
5. 該当する場合、未変化体および/または活性代謝物の曝露レベルを測定する。
6. 同じ方法で腎機能を評価することは、さまざまな臨床研究からのデータを集約する必要がある場合に特に重要である。
7. 曝露-反応関係の分析を行う場合は、腎不全を独立した考察要因と見なされるべきである。
母集団薬物動態分析に関するその他の関連要求事項については、「母集団薬物動態研究の技術ガイドライン」を参照することができる。
(四)透析療法を受けている患者のための研究上の考慮事項
薬物PKに対する透析療法の効果を調べる研究では、対処すべき主な問題は次のとおりである。(1)透析のために用法用量を調整する必要があるかどうか。(2)必要であれば、どう調整するか。(3)透析に関する投与時間。
1. 間欠的透析療法
間欠的透析療法(IHD)、実行できる現在の間欠的透析モードは、低フラックス血液透析(LFHD)、高フラックス透析(HFD)、および血液透析濾過(HDF)である。
IHD研究では、透析期と非透析期(透析間期)の2つの段階がある。各被験者は、2つのシナリオで単回投与を受ける必要がある。(1)透析前に投与され、通常、透析は予想されるTmax投与後の前に開始される。(2)非透析時の予期される曝露を反映する方法で投与すべきである(例えば、透析が完了するまでの一定の時間間隔の後)。
IHDは通常ESRD患者に最もよく用いられる方法であり、そのため、透析研究において最も重要な内容となっている。IHD透析の薬物PKへの影響を他の透析パターンに外推すことは困難である。
2. 持続的透析療法
持続的腎代替療法(CRRT)とは、1日24 h、または24 hに近い時間で持続的に血液を浄化する治療法である。一般的な治療パターンには、連続静脈-静脈血液濾過(CVVH)、連続静脈-静脈血液透析(CVVHD)、連続静脈-静脈血液透析濾過(CVVHDF)がある。治療目標と治療モードの特性に応じて、適切な透析モードを選択する。
CRRT患者に使用される可能性のある集中治療薬に対して、IHD研究の結果は、そのような患者に推奨用量を提供するには不十分な場合がある。したがって、CRRTの薬物PKへの影響を評価し、推奨用法用量を提供することが重要である。このような研究を行う際の挑戦(例えば、患者の血行動態の安定性および血液サンプルの採取の難しさ)を考慮すると、このような状況では、関連するPK情報を収集するために最適なサンプリング時間を設計することが特に重要である。さらに、異なる研究サイト間のCRRT方法の違いによる影響を考慮する必要がある。実行可能な場合、CRRTでの研究結果がある程度の適用性があるように、合理的な設計及び研究の実施が非常に重要である。単回投与研究が最も実行可能な方法である。複数回投与研究を行う場合、患者の状態変化に応じてCRRTの効果を評価できる。1つの方法は、関連する用量情報を得るために、固定された血液流量(QB)および1つまたは2つの一般的に規定された流出流量(QE)を選択することである。
3. 腹膜透析の標準化方案
現在、臨床診療において持続的腹膜透析の方法は以下の数種類がある。持続携帯式腹膜透析(CAPD)、持続的周期的腹膜透析(CCPD)、夜間間欠的腹膜透析(NIPD)、自働化腹膜透析(APD)、このうち、CAPDは現在最も多く選択されている透析方式である。
4. サンプル採取とデータ分析
ESRD患者の非透析(または透析間期)クリアランスを正確に推定するには、薬物と活性代謝物の完全なPK状態を得るために、投与量およびサンプル採取時間を慎重に設計する必要がある。
透析中のクリアランスを決定するために、透析前に血液サンプルを採取し、透析中に適当な間隔で透析器の動脈側と静脈側の血流からサンプルを採取する。異なる時間に体積を記録してサンプルを保持して濃度分析のために使用する。血流、透析中の透析液の流量、及び透析器のメーカーと型番を記録する。血液(透析器に入れるもの)と透析液サンプル中の未変化体と活性代謝物の濃度を測定する。透析液から除去された薬の総量を決定するには、以下の式を用いて透析クリアランスを計算する。

ここに、t0は血液透析の開始時刻、t1は血液透析の終了時刻である。
透析前と透析終了時の血液サンプルも、薬と血漿タンパクへの結合率を測定しなければならない。透析患者に追加投与が必要かどうかを評価するには、投与量に対する透析液中の薬物の割合を計算する必要がある。
同時に、透析後の周囲組織からの薬物の再分布による濃度リバウンドの可能性を考慮し、投与量への影響を確認する必要がある。
(五)薬力学的評価
適切かつ実行可能な場合、薬力学的評価を腎不全の研究に含める必要がある。このような評価は、腎機能の変化がPKの変化なしに(例えば、経口抗凝固剤)薬力学の変化をもたらす場合に特に重要である。薬力学的バイオマーカーの選択は、科学的かつ合理的でなければならない。
四、データ分析
データ分析の目的は、腎不全の患者が用法用量を調整する必要があるかどうかを評価し、必要であれば腎機能の測定結果に基づいて用法用量調整の推奨事項を提供することである。データ分析の過程で、特定の状況に応じて適切なデータ分析の手順や手法を採用することができる。典型的なデータ分析プロセスには以下の手順がある。
1. 薬物および/または活性代謝物のPKパラメータの推定
2. 腎機能指標とPKパラメータ間の相関の数学モデルの確立
3. 腎不全患者の投与量および/または投与頻度調整の必要性の評価
(一)PKパラメータの推定
薬物および/または活性代謝物のPKパラメータを説明及び評価するために、血中濃度データと尿排泄データを分析する。薬物PKパラメーターには、血中濃度-時間曲線(AUC)、最高血中濃度(Cmax)、クリアランス(CL、静脈内投与)または見かけのクリアランス(CL/F、非静脈内投与)、腎クリアランス(CLR)、非腎クリアランス(CLNR)または見かけの非腎クリアランス(CLNR/F)、分布容積(V)または見かけの分布容積(V/F)、終末半減期(t1/2)、遊離型分率(fu)、および透析によるクリアランスCLD(該当する場合)などが含まれる。活性代謝物のPKパラメーターには、AUC、Cmax、CLR、t1/2などが含まれる。PKパラメーターの推定は、ノンコンパートメントモデルおよび/またはコンパートメントモデル解析の方法を使用することができる。
(二)腎機能とPKパラメーターの関係の数学モデルの確立
この手順は、患者の腎機能情報を取得する際に、薬物のin vivo PKパラメーターを予測することができるように、CLCrまたはeGFRのような腎機能とCL、CL/FまたはCLRのようなPKパラメータとの間の相互関係の数学モデルを確立することを目的とする。
腎機能とPKパラメーター間の定量的相関は、連続変数として母集団薬物動態などのモデリングおよびシミュレーション手法を使用して調査できる。この方法は通常、群間のPKと曝露量の差を決定する際に、腎機能を分類変数として扱うよりも優れている。すなわち、腎機能が正常、軽度、中等度、重度の群に分類される。いずれの場合も、薬物PKの違いに影響を与える可能性のあるベースライン共変量(年齢、性別、体重、人種など)を評価する必要がある。同時に、選択したモデルパラメーターとその精度の推定値を計算する。
(三)腎不全患者への推奨用法用量の提供
腎不全患者の用法用量は、腎機能、薬物曝露、効果(有効性と安全性)の関係、及び許容可能な薬物利益とリスクの全体的な認識に基づいて決定されるべきである。治療ウィンドウが広い治験薬の場合、腎機能による薬PKの変化に基づいて、腎不全患者に対する用法用量の調整は必ずしも必要ない。そうでなければ、腎機能が正常な集団の用法用量での効果に見合った曝露に基づき、曝露-反応関係の研究に従って、腎不全患者の用法用量(投与量や投与頻度などを含む)を推奨することができる。
腎不全の患者のために、現在、用法用量を推奨するための様々な研究方法がある。例えば、独立した腎不全患者PKの研究結果については、対照群との比率の記述統計を実行できる。また、モデルシミュレーションを使用して、腎不全患者における治験薬の90%の曝曝区間が、対照群の5%~95%の分位範囲の曝露と同様であることを予測することもできる。さらに、効果のない境界、すなわち、特定の区間内の曝露変化が臨床的意義を生み出すには不十分であることを最初に決定することもできる。用法用量を調整する腎機能限界値は、モデリングおよびシミュレーション手法を用いて決定することができ、この限界値レベルよりも低い場合は、異なる用法用量を与えることが推奨されており、このような場合は、表2に規定されている腎不全分類限界値を参照する必要はない。
場合によっては、さまざまな腎機能推定式を使用して計算された腎不全患者の推奨投与量に有意差がある可能性がある。これは、完全に分析して説明する必要がある。
五、申請資料
申請資料には通常、腎不全患者のPK研究方案、研究報告などの内容が含まれなければならない。腎不全患者のデータが母集団薬物動態研究を通じて評価される場合、「母集団薬物動態研究の技術ガイドライン」を参照して、母集団薬物動態研究報告書を提出することを勧めする。
腎不全患者のPK研究プロトコルには、母集団の選択、サンプルサイズ、投与方法、サンプル採取とデータ分析など、関連する主要な設計が含まれる。サンプルサイズを推定する際の主なパラメーターを設定するための基礎を提供する必要がある。用法用量をサポートするために確立された効果のない境界又はその他の境界値などが含まれている場合は、プロトコルに設定の根拠を提出して説明する必要もある。
腎不全患者のPK研究報告には、PKパラメーター結果および数学モデル構筑のプロセスと結果を含める必要がある。データ表示には、通常、次の方法を含める必要がある。(1)腎機能指標とPKパラメーターの関係をグラフで示す。(2)各腎機能グループ(対照群を含む)の各PKパラメーターおよび対照群パラメータとの比較に関する記述統計データ(例えば、幾何平均およびその比率、算術平均、標準偏差、変動係数、範囲等)を提供する。(3)簡略化されたPK研究設計の場合、重度の腎不全群と対照群の間のPKパラメーターの幾何学的平均比のポイント推定値(および変動係数、信頼区間など)を選択した有意水準で呈する必要がある。腎機能の指標は絶対値mL/minと表示する必要がある。数学モデルは、モデルパラメーターの推定値とその精度と正確度などを提出する必要がある。腎不全患者の用法用量に対する研究結果の影響も、例えば、曝露-反応関係分析を組み合わせて用法用量を調整するための基礎を提供するなど、申請資料に提示する必要がある。
腎不全の被験者で関連する研究が行われない場合は、申請資料の中で科学的根拠を十分に記載し、腎不全が未変化体、活性代謝物および/または主な不活性代謝物のPKに与える影響とリスクの可能性について検討すべきである。
六、添付文書
薬の添付文書は通常、腎不全患者の投薬計画を説明する必要がある。関連する内容の記述は、腎不全患者の研究結果に基づいて作成される。
データが不十分な場合、腎不全患者の治験薬の使用が制限される可能性がある。
腎不全の被験者で関連する研究が実施されていない場合は、添付文書にその旨を記載する必要がある。
七、参考文献
1. U.S. Food and Drug Administration. Guidance for Industry Pharmacokinetics in Patients with Impaired Renal Function – Study Design, Data Analysis, and Impact on Dosing (DRAFT GUIDANCE). 2020.9.
2. European Medicines Agency. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. 2016.7.
3. Matthew A. Ladda and Kerry B. Goralski. The Effects of CKD on Cytochrome P450–Mediated Drug Metabolism, Advances in Chronic Kidney Disease, Vol 23, No 2 (March), 2016: pp 67-75.
4. Delanaye P, Krzesinski J M. Indexing of renal function parameters by body surface area: intelligence or folly? [J]. Nephron Clinical Practice, 2011, 119(4):289-292.
5. Zhang Y, Mehta N, Muhari-Stark E, et al. Pediatric Renal Ontogeny and Applications in Drug Development. J Clin Pharmacol. 2019 Sep;59 Suppl 1:S9-S20.
6. Salvador C L, Tndel C, Rowe A D, et al. Estimating glomerular filtration rate in children: evaluation of creatinine-and cystatin C-based equations[J]. Pediatric Nephrology, 2019, 34(2):301-311.
7. Schwartz G J, Munoz A, Schneider M F, et al. New Equations to Estimate GFR in Children with CKD[J]. Journal of the American Society of Nephrology, 2009, 20(3):629-637.
8. Ying-Chun Ma, Li Zuo, Jiang-Hua Chen, et al. Modified Glomerular Filtration Rate Estimating Equation for Chinese Patients with Chronic Kidney DiseaseJASN October 2006, 17 (10) 2937-2944.
9. Wang Y, PR Jadhav, M Lala, et al. Clarification on Precision Criteria to Derive Sample Size When Designing Pediatric Pharmacokinetic Studies, J Clin Pharmacol, 2012, 52(10):1601-1606.
10. 国家医薬品監督管理局.「母集団薬物動態研究の技術ガイドライン」.2020.
11. 国家医薬品監督管理局.「革新的医薬品の臨床薬理学試験技術ガイドライン」.2021.