

 ホーム
ホーム 最新情報
最新情報 機構紹介
機構紹介 国際交流
国際交流 サイトについて
サイトについて国家食品医薬品監督管理総局(以下「総局」と略称)は、「国務院弁公庁によるジェネリック医薬品品質と治療効果一致性評価実施の意見」(国弁発〔2016〕8号)を実施するために、「ジェネリック医薬品品質と治療効果一致性評価実施関係事項の公告」(総局公告2017年100号)などの書類の要求事項に基づき、「ジェネリック医薬品品質と治療効果一致性評価関係受理審査マニュアル(一致性評価実施必須品目)」と「ジェネリック医薬品品質と治療効果一致性評価関係受理審査マニュアル(国内における同一ラインで生産且つ欧米上市品目)」および関係証票を作成し、2017年9月5日に公表した。
別添:1、ジェネリック医薬品品質と治療効果一致性評価関係受理審査マニュアル(一致性評価実施必須品目)
2、ジェネリック医薬品品質と治療効果一致性評価関係受理審査マニュアル(国内における同一ラインで生産且つ欧米上市品目)
3、ジェネリック医薬品品質と治療効果一致性評価関係証票
別添1
ジェネリック医薬品品質と治療効果一致性評価関係受理審査マニュアル
(一致性評価実施必須品目)
一、適用範囲
国産ジェネリック医薬品と輸入ジェネリック医薬品。
二、資料受領と受理部署
総局行政事項受理サービスと告発苦情処理センター(略称は総局受理と告発処理センター)が資料の受領と申請の受理を行う。
三、申請資料に関する基本的要求事項
(一)申請表の整理
1、種類と部数の要求事項
医薬品補充申請表は3部提出し、そのうちの1部は原本でなければならない。
2、「新版医薬品登録申請表電子版プログラムに関する公告」(総局公告2016年第182号)の要求事項に準じ、申請表の記入と提出は総局が配布したソフトウェアを使用し、新版「医薬品登録申請表電子版プログラム」で作成した電子版または紙質書類を提出しなければならない。また、使用中のバージョンは最新バーションで、電子ファイルのフォーマットはRVT/MRTと確認しなければならない。各ベージのデータ確認コードは必ず一致し、且つ提出する申請表電子版とも一致しなければならない。申請者または登録代行機関は割印を押捺しなければならない。
3、正確に、漏れなく、規則どおりに記入し、手書きまたは修正してはならず、申請表記入関係要求に適合しなければならない。
(二)申請資料の整理
1、申請資料は1式3セットで提出し、そのうちの一部は原本で、残りのコピーは内容が原本と完全に一致しなければならない。また、各セットに関係申請表を入れなければならない。
2、各セットの資料はそれぞれ専用袋に入れ、専用袋は破損を防ぐために、丈夫なクラフト紙でつくられたものを使用するように。また、申請者または登録代行機関は専用袋の表面に申請区分、医薬品名称、第Xセット第X袋(1セット計X袋)、原本かコピー、連絡担当者、電話番号、携帯番号、住所、郵便番号、申請者所属機関名を明記し、袋に捺印しなければならない。
3、申請資料のトップページは目録で、当該目録における資料各項目の番号および名称はいずれも「ジェネリック品質と治療効果一致性評価関係事項の公告」(総局公告2017年第100号。以下「2017年第100号公告」と略称。)などの関係公告で規定された要求事項に準じて記入しなければならない。
4、各セットの各項目申請資料は独立した表紙を持ち、表紙には医薬品名称、資料項目番号、資料項目名称、研究機関およびスタッフ関係項目(適用する場合)、各申請機関名称(登録代行機関の場合も記入)などを記入しなければならない。また、表紙の右上に資料項目番号を明記し、申請機関または登録代行機関が捺印する。
5、資料と書類はいずれもA4紙を使用する。申請資料に添付された図面は見やすくてはっきりし、コピーを使用しないように。フォーマットの要求事項について、「医薬品登録申請資料の体裁と整理規則」(食薬監弁注〔2011〕98号)を参照。
6、申請資料における外国語部分は中国語に翻訳し(参考文献は少なくとも中国語概要および関係部分の全訳を提供)、まずは訳文、次は原文という順番にしなければならない。申請者は責任をもって訳文の正確さを確保しなければならない。原本提出必須な場合、要求どおりの部数を提出しなければならない。
四、申請表審査要点
1、その他特別記載事項:ジェネリック医薬品品質と治療効果一致性評価(以下「一致性評価」と略称。)の申請を行う際に、申請表における特別記載事項で「一致性評価申請、処方と生産手順変更あり」、「一致性評価申請、処方と生産手順変更なし」または「一致性評価免除申請」と明記しなければならない。
2、申請事項分類:医薬品分類および登録分類は「医薬品登録管理規則」別添4における「登録事項」関係要求に準じて「国家食品医薬品監督管理総局審査承認補充申請事項:その他」を選定する。
3、規格:登録申請を行う医薬品が多数の規格がある場合、1通りの規格に1つの表を使う。
4、申請表に記入した情報は医薬品承認書類の関係内容と一致し、処方と生産手順変更の場合は除外。
5、費用支払い情報:処方と生産手順変更の場合、申請者は国に登録申請費用を支払う機関を一つ指定し、「費用支払い機関」のところにチェックを入れなければならない。
6、申請機関の署名と捺印
6.1各申請機関の名称、捺印、法定代表者署名、日時を確認する。
6.2申請機関の捺印はその名称と完全に一致し、国の関係規定に適合し、法的効力を持たなければならない。
6.3法定代表者は申請表に署名、捺印(必要な場合)し、署名は自筆でなければならない。法定代表者以外のものが署名する場合、法定代表者の授権が必要で、署名授権書の原本を提供しなければならない。
五、申請資料審査要点
(一)国産のジェネリック医薬品
1、申請資料要求事項
1.1一致性評価申請
2017年第100号公告における規定に基づいて「化学薬品類ジェネリック医薬品内服製剤品質と治療効果一致性評価申請資料要求事項(試行版)の公表に関する通告」(国家食品医薬品監督管理総局通告2016年第120号。以下は「2016年第120号通告」と略称。)に適合する申請資料を提供しなければならない。そのうち、情報総括表は総局医薬品審査センターのウェブサイトに電子版を提出しなければならない。
1.2一致性評価免除申請
2017年第100号公告第八条に基づいて一致性評価免除を申請する場合、申請者は免除の理由および上市後における処方と生産手順の変更状況を説明すると同時に、再評価実施品目上市済み関係証拠書類を提供しなければならない(変更後の最新版証拠書類を含む)。
2、資料審査の内容
2.1証明書類
2.1.1再評価品目上市済み関係証明書類。変更事項のある最新有効証明書類を含む(あれば、提出のこと)。
医薬品承認証明書類およびその添付文書のコピー。申請事項関係承認書類のすべてを含む。例えば医薬品登録承認書類、補充申請許可書およびその添付文書(医薬品標準、取扱説明書、ラベルのサンプルおよびその別添)。
2.1.2申請者である機関の登録証明書類(営業許可書など)、メーカーの「医薬品生産許可証」および変更事項記載ページ、「医薬品生産品質管理規範」認証証書のコピー。
2.1.3生物的同等性試験が開始する前に、申請者は「医薬品臨床試験情報プラットフォームに関する公告」(総局公告2013年第28号。以下「2013年第28号」と略称。)に基づいて試験項目、臨床試験実施機関、サンプル分析機関、比較用製剤などの情報を総局医薬品審査センターの医薬品臨床試験登録と情報公開プラットフォームに入力しなければならない。
2.1.4比較用製剤の入手ルートおよび関係証明書類。企業が自ら海外から調達した比較用製剤の場合、購買証票、取扱説明書などの資料を提供し、もしくはその他適切な方法で比較用製剤は明記したメーカーの製品を証明しなければならない。
2.1.5 2017年第100号公告の第八条に基づいて一致性評価免除を申し込む場合、一致性評価免除の理由および上市後における処方と生産手順変更状況を説明しなければならない。
2.2技術資料(一致性評価申請関係)
2017年第100号公告の規定に従って2016年120号通告の要求事項を満たす申請資料を提供しなければならない。資料の書式、目録および各項目コードは変更してはならない。関係情報または研究資料がない項目でも、コードと名称を保留し、「関係研究なし」または「不適用」と明記する。
開発企業の中国国内で生産し、上市させた品目は、上市後における重大変更があり、原産国で生産されたものと品質、治療効果で差異がある場合、関係要求に準じて一致性評価を行うのが必須で、国産ジェネリック医薬品の基準に基づいて資料を提出しなければならない。
(二)輸入ジェネリック医薬品
1、申請資料要求事項
1.1一致性評価申請
2017年第100号公告における規定に基づいて2016年第120号通告の要求に適合する申請資料を提供しなければならない。医薬品規制調和国際会議(ICH)が規定したコモン?テクニカル?ドキュメント(CTD)の1式、および2016年120号通告で要求された「概要」と「体外評価」の部分を提出することも認められる。そのうち、情報総括表は総局医薬品審査センターのウェブサイトに電子版を提出しなければならない。
1.2一致性評価免除申請
申請者が2017年第100号公告第八条に基づいて一致性評価免除申請を行う場合、一致性評価免除申請理由および上市後における処方と生産手順変更状況の説明を行い、再評価品目上市済み関係証明書類も提供しなければならない(変更関係最新版有効証明書類があれば、提供必須)。
2、資料審査の内容
2.1証明書類
2.1.1再評価品目上市済み関係証明書類。変更事項のある最新版有効証明書類を含む(あれば、提出)。
処方と生産手順を変更した場合、その生産国または地域の医薬品管理機関が発行した医薬品変承認証明書類、公証文書およびその中国語訳。書式はWHOが推奨するものを採用しなければならない。その他書式の書類は、所在国の公証機関および中国大使館の承認が必要となる。生産国または地域における医薬品管理機関が関係証明書類を発行できない場合、現地の法律と法規に基づき、説明を行うことができる。
2.1.2海外製薬企業の駐中国事務所が登録を代行する場合、「外国企業駐中国代表機構登録証」のコピーを提供しなければならない。海外製薬企業が中国の会社か機構に登録申請の代行を依頼する場合、委託書、公証文書およびその中国語訳、代理者の「登記簿」謄本を提供しなければならない。
2.1.3生物的同等性試験が開始する前に、申請者は2013年第28号公告に基づいて試験項目、臨床試験実施機関、サンプル分析機関、比較用製剤などの情報を総局医薬品審査センターの医薬品臨床試験登録と情報公開プラットフォームに入力しなければならない。
2.1.4比較用製剤の入手ルートおよび関係証明書類。企業が自ら海外から調達した比較用製剤の場合、購買証票、取扱説明書などの資料を提供し、もしくはその他適切な方法で比較用製剤のメーカーを証明しなければならない。
2.1.5 2017年第100号公告の第八条に基づいて一致性評価免除を申し込む場合、一致性評価免除の理由および上市後における処方と生産手順変更状況を説明しなければならない。
2.2技術資料(一致性評価申請関係)
2017年第100号公告の規定に従って2016年120号通告の要求事項を満たす申請資料を提供しなければならない。医薬品規制調和国際会議(ICH)が規定したコモン?テクニカル?ドキュメント(CTD)の1式、および2016年120号通告で要求された「概要」と「体外評価」の部分を提出することも認められる。
資料の書式、目録および各項目コードは変更してはならない。関係情報または研究資料のない項目でも、コードと名称を保留し、「関係研究なし」または「不適用」と明記する。ICHが規定したCTDの1式を提出する場合、ICH M4の1式を全部提出すると同時に、2016年120号通告における資料項目に準じて作成された資料の目録と索引を提供しなければならない。
六、資料受領と申請受理審査決定
(一)資料受領と申請受理
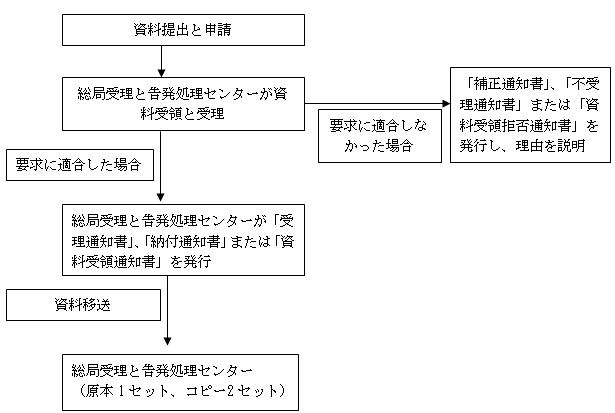
1、資料受領通知書と申請受理通知書:形式審査実施条件に適合すると判断した場合、「ジェネリック品質と治療効果一致性評価申請資料形式審査表」を記入し、「資料受領通知書」(総局行政許可資料受領専用印押捺済み)と「申請受理通知書」(総局行政許可受理専用印押捺済み)を1式2部で発行し、申請者に1部渡し、もう1部を資料として保存する。
受領番号と受理番号発行原則:処方と生産手順が不変で、一致性評価の実施または免除を申請する場合、CYHBXX(最後の2桁は年)XXXXX国(4からはじまり、5桁の自然順番号)或JYHBXX(最後の2桁は年)XXXXX国(4からはじまり、5桁の自然順番号)の受領番号が発行される。処方と生産手順変更に関する一致性評価の場合、CYHBXX(最後の2桁は年)XXXXX国(5からはじまり、5桁の自然順番号)或JYHBXX(最後の2桁は年)XXXXX国(5からはじまり、5桁の自然順番号)が発行される。
2、納付通知書:処方と生産手順不変、もしくは一致性評価免除申請の場合、費用を支払う必要はない。処方と生産手順が変更した場合、医薬品登録補充申請(技術審査必須)の基準に準じて費用を支払わなければならない。
(二)補正
申請資料不備または法定の形に適合していない場合、一括で申請者に補正すべきことを告知し、「補正通知書」を発行する。
(三)資料受領拒否と申請不受理
要求条件に適合していないと判断した場合、「資料受領拒否通知書」または「申請不受理通知書」を発行し、理由も説明する。
七、その他
その他関係事項については、2017年100号公告、「ジェネリック医薬品品質と治療効果一致性評価実施品目分類ガイドライン」(総局通告2017年第49号)、「『国務院弁公庁によるジェネリック医薬品品質と治療効果一致性評価の意見』実施関係事項の公告」(総局公告2016年第106号)と「ジェネリック医薬品品質と治療効果一致性評価手順の公告」(総局公告2016年第105号)などに基づいて実施するものとする。
八、受理フロー図

九、別添
ジェネリック医薬品品質と治療効果一致性評価申請資料形式審査表
|
□一致性評価申請(□処方と生産手順不変 □処方と生産手順不変) □一致性評価免除申請 |
|||||
|
第一部分 申請資料書式と整理関係要求 |
|||||
|
1、申請資料3セット。原本は少なくとも1セット。 |
□はい |
□いいえ |
|
||
|
2.資料項目目録は総局2017年第100号公告など関係文書の要求に準じて並べ方を決める。 |
□あり |
□なし |
|
||
|
3.申請資料における外国語部分は中国語に翻訳しなければならない(参考文献は少なくとも中国語概要および関係部分の全訳を提供)。 |
□はい |
□いいえ |
|
||
|
第二部分 申請表審査 |
|||||
|
1、「医薬品補充申請表」(通常) |
|||||
|
1.1 1式3部。原本は少なくとも1部。 1.2 法定代表者署名/申請者公印押捺/割印 |
□はい □あり |
□いいえ □なし |
|
||
|
2.「医薬品補充申請表」記入状況 |
|||||
|
2.1申請事項分類適正さの確認 |
□はい |
□いいえ |
|
||
|
2.2その他特別記載事項:「一致性評価申請、処方と生産手順変更あり」、「一致性評価申請、処方と生産手順変更なし」または「一致性評価免除申請」 |
□あり |
□なし |
|
||
|
2.3医薬品関係情報:要求に準じて医薬品情報を記入したかどうか |
□あり |
□なし |
|
||
|
2.4申請者情報は正確かどうか |
□はい |
□いいえ |
|
||
|
2.5委託研究か生産機関 |
□あり □不適用 |
□なし |
|
||
|
2.6費用納付機関(処方と生産手順変更の場合) |
□あり |
□なし |
|
||
|
第三部分 証明書類関係要求 |
|||||
|
1.再評価品目上市済み関係証明書類 |
□あり |
□なし |
|
||
|
2.医薬品臨床試験登録情報 |
□あり □不適用 |
□なし |
|
||
|
3.比較用製剤の入手ルートおよび関係証明書類。企業が自ら海外から調達した比較用製剤の場合、購買証票、取扱説明書などの資料を提供し、もしくはその他適切な方法で比較用製剤のメーカーを証明しなければならない。 |
□あり □不適用 |
□なし |
|
||
|
4.総局2017年第100号公告第八条に基づいて一致性評価免除を申請する場合、免除の理由および上市後における処方と生産手順変更状況を説明しなければならない。 |
□あり □不適用 |
□なし |
|
||
|
5.上述した資料のほか、国産ジェネリック医薬品の場合は以下の証明書類を提出しなければならない。 「企業法人営業許可証」など 「医薬品生産許可証」における製品範囲は本剤型を含むかどうか 関係剤型の「医薬品生産品質管理規範」認証証書 |
□不適用 □あり □はい □あり |
□なし □いいえ □なし |
|
||
|
6.上述した資料のほか、輸入ジェネリック医薬品の場合は以下の証明書類を提出しなければならない。 代理申請委託文書、「企業法人営業許可証」または「外国企業中国駐在代表機関登記簿」のコピー |
□不適用 □あり |
□なし |
|
||
|
第四部分 申請資料要求 |
|||||
|
1.基本要求事項 |
|||||
|
1.1総局2017年第100号公告などの文書における要求に基づいて申請資料を提出する。 |
□はい |
□いいえ |
|
||
|
1.2医ICHが規定しCTDの1式、および2016年120号通告で要求された「概要」と「体外評価」の部分を提出し、2016年120号通告における資料項目に準じて作成された資料の目録と索引を提供しなければならない。 |
□あり □不適用 |
□なし |
|
||
|
2.申請資料と目録内容が一致しているかどうか確認 |
□はい |
□いいえ |
|
||
|
結論:
担当者: 時間: |
|||||
別添2
ジェネリック医薬品品質と治療効果一致性評価受理審査マニュアル
(国内の同一ラインで生産し、且つ欧米と日本で上市した品目)
一、適用範囲
中国国内企業が生産し、EU、アメリカまたは日本で上市が許可されたジェネリック医薬品で、中国国内でも上市済みで、同一生産ライン、同一処方と生産手順を使用した品目。
中国国内企業が生産し、EU、アメリカまたは日本で上市が許可されたジェネリック医薬品で、中国国内でも上市済みで、異なる生産ラインまたは異なる処方と生産手順を使用した品目。
注:中国国内企業が生産し、EU、アメリカまたは日本で上市したものの、中国国内では未上市である場合、総局に上市申請を提出するべきである。
二、資料受領と申請受理部署
総局行政事項受理サービスと告発苦情処理センター(略称は総局受理と告発処理センター)が資料の受領と申請の受理を行う。
三、申請資料に関する基本的要求事項
(一)申請表の整理
1、種類と部数の要求事項
医薬品補充申請表は3部提出しなければならない。そのうちの1部は原本でなければならない。
2、「新版医薬品登録申請表電子版プログラムに関する公告」(総局公告2016年第182号)の要求事項に準じ、申請表の記入と提出は総局が配布したソフトウェアを使用し、新版「医薬品登録申請表電子版プログラム」で作成した電子版または紙質書類を提出しなければならない。また、使用中のバージョンは最新バーションで、電子ファイルのフォーマットはRVT/MRTと確認しなければならない。各ベージのデータ確認コードは必ず一致し、且つ提出する申請表電子版とも一致しなければならない。申請者または登録代行機関はページング?シールという形で各ベージに捺印しなければならない。
3、正確に、漏れなく、規則どおりに記入し、手書きまたは修正してはならず、申請表記入関係要求に適合しなければならない。
(二)申請資料の整理
1、申請資料は1式3セットで提出し、そのうちの一部は原本で、残りのコピーは内容が原本と完全に一致しなければならない。また、各セットに関係申請表を入れなければならない。
2、各セットの資料はそれぞれ専用袋に入れ、専用袋は破損を防ぐために、丈夫なクラフト紙でつくられたものを使用するように。また、申請者または登録代行機関は専用袋の表面に申請区分、医薬品名称、第Xセット第X袋(1セット計X袋)、原本かコピー、連絡担当者、電話番号、携帯番号、住所、郵便番号、申請者所属機関名を明記し、袋に捺印しなければならない。
3、申請資料のトップページは目録で、当該目録における資料各項目の番号および名称はいずれも「ジェネリック品質と治療効果一致性評価関係事項の公告」(総局公告2017年第100号。以下「2017年第100号公告」と略称。)などの関係公告で規定された要求事項に準じて記入しなければならない。
4、各セットの各項目申請資料は独立した表紙を持ち、表紙には医薬品名称、資料項目番号、資料項目名称、研究機関およびスタッフ関係項目(適用する場合)、各申請機関名称(登録代行機関の場合も記入)などを記入しなければならない。また、表紙の右上に資料項目番号を明記し、申請機関または登録代行機関が捺印する。
5、資料と書類はいずれもA4紙を使用する。申請資料に添付された図面は見やすくてはっきりし、コピーを使用しないように。フォーマットの要求事項について、「医薬品登録申請資料の体裁と整理規則」(食薬監弁注〔2011〕98号)を参照。
6、申請資料における外国語部分は中国語に翻訳し(参考文献は少なくとも中国語概要および関係部分の全訳を提供)、まずは訳文、次は原文という順番にしなければならない。申請者は責任をもって訳文の正確さを確保しなければならない。原本提出必須な場合、要求どおりの部数を提出しなければならない。
四、申請表審査要点
1、その他特別記載事項:一致性評価の申請を行う際に、申請表における特別記載事項で「同じライン生産一致性評価申請、生産ラインと手順、処方変更あり」または「同じライン生産一致性評価申請、生産ラインと手順、処方変更なし」と明記しなければならない。
2、申請事項分類:医薬品分類および登録分類は「医薬品登録管理規則」別添4における「登録事項」関係要求に準じて「国家食品医薬品監督管理総局審査承認補充申請事項:その他」を選定する。
3、規格:登録申請を行う医薬品が多数の規格がある場合、1通りの規格に1つの表を使う。
4、補充申請の理由:生産場所から生産ラインまでの詳細情報を提供し、国内外で上市した当該製品は同一生産ラインで生産され、原料と添加剤の由来と品質、処方と生産手順、品質制御関係要求なども国内外で不変であると誓約する必要がある。
5、申請表に記入した情報は医薬品承認書類の関係内容と一致しなければならない。処方と生産手順変更の場合は除外。
6、費用支払い情報:処方と生産手順変更の場合、申請者は国に登録申請費用を支払う機関を一つ指定し、「費用支払い機関」のところにチェックを入れなければならない。
7、申請機関の署名と捺印
7.1各申請機関の名称、捺印、法定代表者署名、日時を確認する。
7.2申請機関の捺印はその名称と完全に一致し、国の関係規定に適合し、法的効力を持たなければならない。
7.3法定代表者は申請表に署名、捺印(必要な場合)し、署名は自筆でなければならない。法定代表者以外のものが署名する場合、法定代表者の授権が必要で、署名授権書の原本を提供しなければならない。
五、申請資料審査要点
1、申請資料要求事項
2017年第100号公告第九条と第十三条第(一)項に準じて申請資料を提出しなければならない。そのうち、情報総括表は国家食品医薬品監督管理総局医薬品審査センターのウェブサイトで電子版を提出しなければならない。
2、資料審査の内容
2.1証明書類
2.1.1再評価品目上市済み関係証明書類。変更事項のある最新有効証明書類(あれば、提出)。
医薬品承認証明書類およびその添付文書のコピー。申請事項関係承認書類のすべてを含む。例えば医薬品登録承認書類、補充申請許可書およびその添付文書(医薬品標準、取扱説明書、ラベルのサンプルおよびその別添)。(あれば、提出)。
海外上市許可証明書類。具体的には、申請者はEU、アメリカまたは日本医薬品管理機関が医薬品上市を許可する証明書類、関係変更証明書類(あれば、提出)、且つ上記国と地域の医薬品管理機関が医薬品市販を許可する証明書類(あれば、提出)を提供しなければならない。
2.1.2申請者である機関の登録証明書類(営業許可書など)、メーカーの「医薬品生産許可証」および変更事項記載ページ、「医薬品生産品質管理規範」認証証書のコピー。
2.1.3原薬、添加剤と医薬品直接接触包装資材の由来および関係証明書類。
原薬、添加剤と医薬品直接接触包装資材の由来が合法的であると証明する書類を提出できない場合、メーカーは原薬、添加剤と医薬品直接接触包装資材がEU、アメリカまたは日本で合法的に使用したという声明書を提出するとう同時に(製剤メーカーが代わりに提出可)、関係技術文書を提出できない理由も説明しなければならない。
2.2技術資料2017年第100号公告第九条と第十三条第(一)項の要求事項を満たす申請資料を提供しなければならない。
そのうち、医薬品規制調和国際会議(ICH)が規定したコモン?テクニカル?ドキュメント(CTD)の1式、ICH M4の1式を全部提出すると同時に、「化学薬品類ジェネリック医薬品内服固体製剤品質と治療効果一致性評価申請資料要求事項(試行版)の発表に関する通告」(国家食品医薬品監督管理総局通告2016年第120号。以下「2016年120号通告」と略称。)における資料項目に基づいて作成された詳細目録と索引を提供しなければならない。海外上市申請を行う際に提出した生物学的同等性試験、薬学的研究のデータを提出する場合、それらのデータはEU、アメリカまたは日本の監管機関に提出した上市申請のためのデータで、中国の現行技術指導原則に準じ、総局による査察を受けなければならない。
六、資料受領と申請受理審査決定
(一)資料受領と申請受理
1、資料受領通知書と申請受理通知書:形式審査実施条件に適合すると判断した場合、「ジェネリック品質と治療効果一致性評価申請資料形式審査表」を記入し、「資料受領通知書」(総局行政許可資料受領専用印押捺済み)と「申請受理通知書」(総局行政許可受理専用印押捺済み)を1式2部で発行し、申請者に1部渡し、もう1部を資料として保存する。
受領番号と受理番号発行原則:生産ライン処方手順が不変である場合、CYHBXX(最後の2桁は年)XXXXX国(4からはじまり、5桁の自然順番号)受領番号が発行される。生産ライン処方手順変更に関する一致性評価の場合、CYHBXX(最後の2桁は年)XXXXX国(5からはじまり、5桁の自然順番号)という受理番号が発行される。
2、納付通知書:生産ライン処方手順が不変である場合、費用を支払う必要はない。生産ライン処方手順が変更した場合、医薬品登録補充申請(技術審査必須)の基準に準じて費用を支払わなければならない。
(二)補正
申請資料不備または法定の形に適合していない場合、一括で申請者に補正すべきことを告知し、「補正通知書」を発行する。
(三)資料受領拒否と申請不受理
要求条件に適合していないと判断した場合、「資料受領拒否通知書」または「申請不受理通知書」を発行し、理由も説明する。
七、その他
その他関係事項については、2017年100号公告、「ジェネリック医薬品品質と治療効果一致性評価実施品目分類ガイドライン」(総局通告2017年第49号)、「『国務院弁公庁によるジェネリック医薬品品質と治療効果一致性評価の意見』実施関係事項の公告」(総局公告2016年第106号)と「ジェネリック医薬品品質と治療効果一致性評価手順の公告」(総局公告2016年第105号)などに基づいて実施するものとする。
八、受理フロー図
九、別添
ジェネリック医薬品品質と治療効果一致性評価申請資料形式審査表
(国内の同一ラインで生産し、且つ欧米と日本で上市した品目)
|
□処方と生産手順不変 □処方と生産手順変更 |
|||||
|
第一部分 申請資料書式と整理関係要求 |
|||||
|
1、申請資料3セット。原本は少なくとも1セット。 |
□はい |
□いいえ |
|
||
|
2.資料項目目録は総局2017年第100号公告など関係文書の要求に準じて並べ方を決める。 |
□あり |
□なし |
|
||
|
3.申請資料における外国語部分は中国語に翻訳しなければならない(参考文献は少なくとも中国語概要および関係部分の全訳を提供)。 |
□はい |
□いいえ |
|
||
|
第二部分 申請表審査 |
|||||
|
1、「医薬品補充申請表」(通常) |
|||||
|
1.1 1式3部。原本は少なくとも1部。 1.2 法定代表者署名/申請者公印押捺/割印 |
□はい □あり |
□いいえ □なし |
|
||
|
2.「医薬品補充申請表」記入状況 |
|||||
|
2.1申請事項分類適正さの確認 |
□はい |
□いいえ |
|
||
|
2.2その他特別記載事項:「同一ライン生産で一致性評価申請、処方と生産手順変更あり」、「同一ライン生産で一致性評価申請、処方と生産手順変更なし」 |
□あり |
□なし |
|
||
|
2.3医薬品関係情報:要求に準じて医薬品情報を記入したかどうか |
□あり |
□なし |
|
||
|
2.4申請者情報は正確かどうか |
□はい |
□いいえ |
|
||
|
2.5委託研究か生産機関 |
□あり □不適用 |
□なし |
|
||
|
2.6費用納付機関(処方と生産手順変更の場合) |
□あり |
□なし |
|
||
|
第三部分 証明書類関係要求 |
|||||
|
1.再評価品目上市済み関係証明書類 |
□あり |
□なし |
|
||
|
2.医薬品臨床試験登録情報 |
□あり □不適用 |
□なし |
|
||
|
3.比較用製剤の入手ルートおよび関係証明書類。企業が自ら海外から調達した比較用製剤の場合、購買証票、取扱説明書などの資料を提供し、もしくはその他適切な方法で比較用製剤のメーカーを証明しなければならない。 |
□あり □不適用 |
□なし |
|
||
|
4.総局2017年第100号公告第八条に基づいて一致性評価免除を申請する場合、免除の理由および上市後における処方と生産手順変更状況を説明しなければならない。 |
□あり □不適用 |
□なし |
|
||
|
5.上述した資料のほか、国産ジェネリック医薬品の場合は以下の証明書類を提出しなければならない。 「企業法人営業許可証」など 「医薬品生産許可証」における製品範囲は本剤型を含むかどうか 関係剤型の「医薬品生産品質管理規範」認証証書 |
□不適用 □あり □はい □あり |
□なし □いいえ □なし |
|
||
|
6.上述した資料のほか、輸入ジェネリック医薬品の場合は以下の証明書類を提出しなければならない。 代理申請委託文書、「企業法人営業許可証」または「外国企業中国駐在代表機関登記簿」のコピー |
□不適用 □あり |
□なし |
|
||
|
第四部分 申請資料要求 |
|||||
|
1.基本要求事項 |
|||||
|
1.1総局2017年第100号公告などの文書における要求に基づいて申請資料を提出する。 |
□はい |
□いいえ |
|
||
|
1.2医ICHが規定しCTDの1式、および2016年120号通告で要求された「概要」と「体外評価」の部分を提出し、2016年120号通告における資料項目に準じて作成された資料の目録と索引を提供しなければならない。 |
□あり □不適用 |
□なし |
|
||
|
2.申請資料と目録内容が一致しているかどうか確認 |
□はい |
□いいえ |
|
||
|
結論:
担当者: 時間: |
|||||
(出所:CFDAサイト2017-09-05)