

 首页
首页 最新动态
最新动态 机构简介
机构简介 国际交流
国际交流 关于我们
关于我们一、药品批准生产上市情况
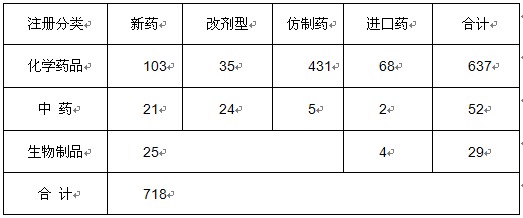
2011年,共批准药品注册申请718件。其中批准境内药品注册申请644件,批准进口74件。
在644件境内药品注册申请中,化学药品569件,占88.4%;中药50件,占7.8%;生物制品25件,占3.8%。
从注册分类看,境内药品注册申请中,新药149件,占22.9%;改剂型59件,占9.3%;仿制药436件,占67.7%。与2010年相比较,批准化学药品仿制药品的数量减少,批准新药的数量增加,其中,1.1类化学药品共批准10件,相比2009年及2010年有显著增长。
表1 2011年批准的药品情况
注:1. 表中数据以受理号计,受理号系申请人提出的一件申请事项的编号。对各申请企业的原料药、制剂、制剂不同规格分别予以编号。
2. 表中新药系根据《药品注册管理办法》规定按照新药管理的药品。化学药品新药包括化学药品注册分类1-4,中药新药包括中药、天然药物注册分类1-7。
3. 表中化学药品改剂型为化学药品注册分类5,中药改剂型为中药、天然药物注册分类8。
4. 表中化学药品仿制药为化学药品注册分类6,中药仿制药为中药、天然药物注册分类9。
5. 生物制品不进行分类。
图1 2009~2011年批准国产药品的对比
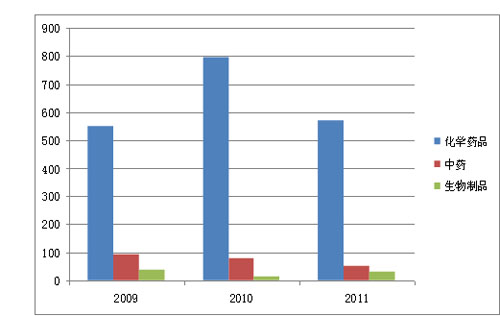
表2 2011年批准的化学药品新药分布

注:“其他”指按《药品注册管理办法》(2005年版)分类申报的一、二、三、四类药品。数量以受理号计。
表3 2011年批准的中药新药分布

注:“其他”指按《药品注册管理办法》(2005年版)分类申报的三类药品。数量以受理号计。
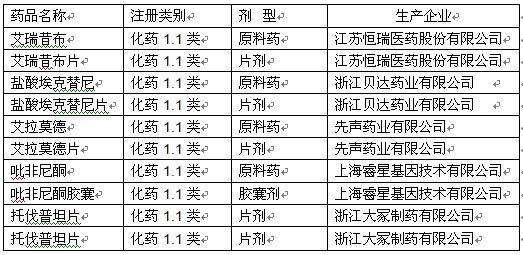
表4 2011年批准的1.1类新药
二、药物临床研究批准情况
2011年,共批准621个注册申请开展临床研究。其中39个为注册分类1类的化学药品注册申请,110件为国际多中心临床研究申请。批准进入临床试验的药物,既涵盖在我国疾病谱中占重要位置的常见疾病和多发疾病,如肿瘤、心血管病等的治疗药物,也包括了社会影响度高的一些罕见性疾病的治疗药物。对于符合《新药注册特殊审批管理规定》要求的,按照特殊审批程序开展审评审批,促进药物研究进程。
表5 2011年药物临床研究批准情况
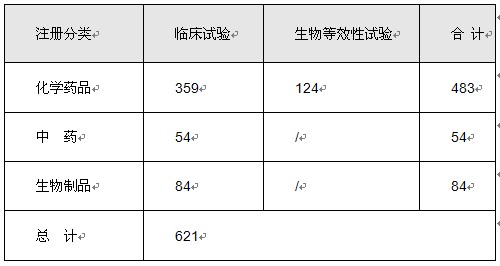
注:以受理号计
三、注册申请受理情况
2011年,国家食品药品监督管理局共受理药品新注册申请3620件,其中境内药品注册申请2913件,进口药品注册申请707件。在2913件境内药品注册申请中,新药1078件,占37.0%;改剂型169件,占5.8%;仿制药1666件,占57.2%。与2010年相比,境内新注册申请量增加20%,新药申报量与2010年相比略有增加,改剂型及仿制药申报量与2010年相比增加34%。
表6 2011年药品新注册受理情况表
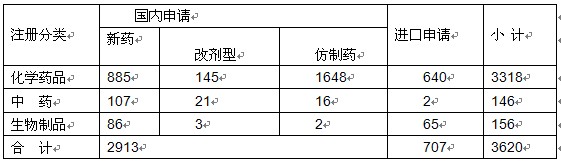
注:以受理号计。
表7 2011年药品补充申请受理情况表
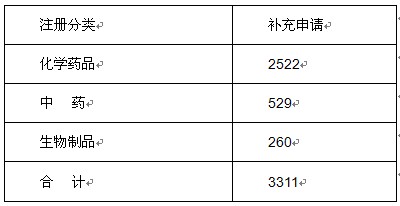
图2 2005至2011年药品注册申请数量变化趋势
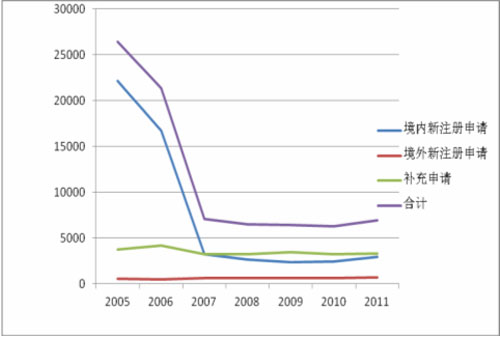
注:以上数据以受理号计;补充申请包括境内、境外的补充申请。
四、批准重要治疗领域药品品种情况
1.临床急需药品
雷珠单抗注射液是抑制血管内皮生长因子的重组抗体药物,用于治疗老年性湿性黄斑变性,是临床急需药品之一。2011年,批准了该药品进口,满足了我国患者的用药需求。
特发性肺纤维化属于罕见病,严重影响肺功能,预后效果差,目前尚无有效治疗药物。2011年,批准了国内首个吡非尼酮胶囊生产,使我国患者能尽早获得有效的治疗药物。
为缓解凝血因子类血液制品供应紧缺局面,批准了重组人凝血因子VIII或IX因子融合蛋白开展临床试验,为血友病患者提供参与临床用药及治疗的机会。
2.预防用生物制品
批准了我国自主研发的重组人戊型肝炎疫苗生产,这是全球首家获得批准的戊型肝炎疫苗,为戊型肝炎流行区高危人群提供了预防途径。
批准了我国自主研发的Sabin株脊髓灰质炎灭活疫苗进入Ⅲ期临床试验。该疫苗对于防止继发于口服脊髓灰质炎减毒活疫苗之后因突变、免疫缺陷等导致的脊髓灰质炎相关病例以及彻底消灭脊髓灰质炎疾病具有十分重要的意义。
为有效应对手足口病对公共卫生健康的威胁,继2010年启动特殊审批程序批准国内3家企业申报的肠道病毒71型(EV71)灭活疫苗进入I期、II期临床试验后,2011年在国家食品药品监督管理局制定的临床试验联合工作机制下,国家食品药品监督管理局药品审评中心对临床试验具体实施给予了技术指导,保障了Ⅲ期临床试验的稳步、有序、顺利开展。
3 . 特殊人群用药
批准了盐酸多奈哌齐口腔崩解片的国内生产及进口上市。该口腔崩解片可解决老年性痴呆症患者的用药顺应性问题,对减缓老年性痴呆症进展具有一定意义。
4 . 治疗类风湿性关节炎和骨关节炎药品
目前,用于类风湿关节炎的慢作用药有限,大多为说明书外的经验用药,且不良反应严重。批准了艾拉莫德片和艾瑞昔布片在全球首家上市,两者均为我国自主知识产权、并列入新药创制重大专项支持的药物。艾拉莫德片用于治疗类风湿关节炎药物,其作用机理趋向于慢作用药,有望缓解疾病病程,现有资料提示不良反应相对较小。艾瑞昔布片为非甾体抗炎药,通过抑制环氧酶(COX)发挥镇痛作用,用于缓解骨关节炎的疼痛症状。
5. 治疗HIV感染的药品
批准了首家国产复方制剂奈韦拉平司他拉米双夫定片(Ⅱ)的生产。该产品是参照WHO推荐的抗HIV治疗(成人及青少年)治疗方案"司他夫定或齐多夫定+拉米夫定+奈韦拉平或依法韦伦"组成的复方制剂。
批准了齐多夫定片和齐多拉米双夫定片。两者均为经典的核苷类逆转录酶抑制剂(NRTIs),是WHO和我国艾滋病治疗指南中抗病毒治疗的一线标准用药,它们的获批,提高了治疗药物的供给性,符合国家传染病防控策略。
在前期预防艾滋病研究结果基础上,批准了艾滋病疫苗(核酸疫苗与重组痘苗联合使用)增加用于治疗成人艾滋病的临床试验研究,以考察疫苗在HIV感染者中诱导体液和细胞免疫、与抗逆转录病毒药物联合应用增强特异性免疫反应、以及控制病毒复制的作用。
6. 治疗乙肝的药品
富马酸替诺福韦二吡呋酯片是国际上公认的治疗乙肝的核苷(酸)类似物之一。2011年,批准了该品种针对乙肝适应症进行临床试验。此外,还批准国内外均未上市、用于乙肝治疗的富马酸替诺福韦双特戊酯片进行I期临床试验。
7. 治疗疟疾的药品
批准国产首家青蒿琥酯阿莫地喹片生产。该品种是全球抗疟委员会、WHO等组织确定的抗疟首选药,可避免抗疟药物抗性下降和遏制疟原虫耐药性,从而更有效地控制和治疗疟疾。
8 . 利尿药
批准国产首家托伐普坦片生产。该品种是非肽性选择性精氨酸加压素V2受体拮抗剂,通过抑制加压素与肾集合管的V2受体结合,促肾脏排水但不增加排钠,对各种原因引起的非低容量性低钠血症具有明显提高血钠浓度的作用。
9. 抗凝药
批准了首家注射用比伐芦定生产上市,比伐芦定做为凝血酶直接抑制剂,通过与游离及血栓上凝血酶的催化位点和阴离子外结合位点特异结合起抑制作用,用于成人择期经皮冠状动脉介入治疗(PCI)。
10. 治疗骨质疏松的药品
批准了特立帕肽注射液进口。特立帕肽注射液是全球第一个上市的人甲状旁腺激素(PTH)(1-34)生物制品,它直接作用于成骨细胞,刺激骨骼形成,适用于有骨折高发风险的绝经后妇女骨质疏松症的治疗,为绝经后妇女骨质疏松症提供了新的治疗手段。
11. 治疗糖尿病的药品
批准了沙格列汀片和利拉鲁肽注射液进口。沙格列汀片属二肽基肽酶4(DPP4)竞争性抑制剂,不仅可以单独使用,还可与盐酸二甲双胍联用,进一步改善糖尿病患者的血糖控制。利拉鲁肽注射液为国内首家上市的GLP-1类似物的生物制剂,其特点为具有长效作用,可一天给药一次。两者均以葡萄糖浓度依赖的模式刺激胰岛素的分泌,低血糖的发生率相对较低,为 = 2 * ROMAN II型糖尿病患者提供了新的治疗手段。
12. 治疗精神类疾病药品
批准了棕榈酸帕利哌酮注射液进口,该药品属于已上市精神类药物中唯一的1个月长效治疗制剂,有利于提高精神分裂症患者临床用药的依从性及防止疾病的复发。批准了阿戈美拉汀片进口,该品种为新型抗抑郁药,研究结果显示,该药不妨碍睡眠结构,对抑郁患者的睡眠有一定改善作用。
13. 抗肿瘤药
目前,对于伊马替尼耐药或不耐受的慢性髓细胞白血病患者的治疗手段有限。2011年,批准了达沙替尼片进口,用于伊马替尼耐药或不耐受的费城染色体阳性(Ph+)慢性髓细胞白血病慢性期、加速期和急变期(急粒变和急淋变)成年患者的治疗。该品种为继尼洛替尼之后另一个用于伊马替尼耐药和不耐受的慢性髓细胞白血病患者的药品,为此类患者提供了更多的治疗手段。
14. 非氟利昂吸入式气雾剂
按照我国履行《蒙特利尔国际公约》的有关工作计划,为加快药用气雾剂用氟利昂的淘汰,按照吸入式气雾剂抛射剂替代的技术要求,批准了非氟利昂硫酸沙丁胺醇气雾剂的生产和临床研究,保证了哮喘、慢性阻塞性肺部疾病的一线治疗用药。同时,批准了非氟利昂丙酸倍氯米松气雾剂的生产,推进了我国药用吸入式气雾剂淘汰氟利昂的工作进展。
15. 中药
根据《中药、天然药物注射剂研究基本技术要求》,经过严格的风险效益评估,批准了2个物质基础相对明确,质量可控程度较高的有效部位中药注射剂生产。批准的其它中药新药主要包括:消化系统疾病的症状改善用药(缬草提取物胶囊、连苏胶囊等)、骨性关节炎和类风湿改善症状用药(丹参通络膏等)、小儿遗尿(小儿益麻颗粒)、多动症(小儿黄龙颗粒)、前列腺炎(丹益片等)、前列腺增生(灵泽片)等症状改善用药,感冒(荆感胶囊)等病毒感染为主的自限性疾病等。
五、 加强药品注册管理的工作举措
2011年是实施国家药品安全“十二五”规划的开局之年,药品注册管理工作积极践行科学监管理念,继续以提高“质量和效率”为中心,以加强药物临床试验监管和规范药品注册管理行为为重点,稳步推进体制机制改革和法规建设,全面加强药物研究全过程的管理,不断完善药品标准管理工作。
1. 完善药品注册管理法规和技术指导原则体系
进一步完善技术指导原则体系。为加强药物临床试验生物样本分析实验室的管理,提高生物样本分析数据的质量和管理水平,发布实施《药物临床试验生物样本分析实验室管理指南(试行)》。为科学规范和指导中药变更研究和中药、天然药物临床研究工作,发布实施了《已上市中药变更研究技术指导原则(一)》、《中药、天然药物治疗冠心病心绞痛临床研究技术指导原则》、《中药、天然药物治疗女性更年期综合征临床研究技术指导原则》等指导原则。
不断规范药品技术审评工作。发布了《审评原则和程序》、《药品审评中心技术审评决策路径管理规范(试行)》和《药品审评中心审评任务管理规范(试行)》,明确了技术审评的决策机制和模式,保证技术审评的质量与效率。
2. 持续推进药品审评审批的科学化
为进一步保证审评质量,提高审评效率,国家食品药品监督管理局药品审评中心调整了内部机构(见附图3),创新药和仿制药分别由不同的部门进行审评;完善了审评模式,按照审评任务分类和风险等级分类设定审评程序,将药品注册申请(含补充申请)按照六个通路开展审评。创新药审评时限进一步缩短,批准临床试验的排队等待时间由过去的9-10个月减少至5. 8个月,批准生产的审评时间平均为10-11个月。建立了职业化、专业化的审评职务体系,强化主审人员和审评团队负责,健全了审评纠错、学术监督和质量评价机制,进一步保证了审评工作质量。
继续通过重大专项专题会、创新品种沟通交流会、专家咨询会议,以及第三方验证、专家票决等方式,保证技术审评工作的科学性和公正性。继续加大公开透明力度,尊重申请人和社会公众的知情权并接受社会监督,将新注册申请的审评计划,按照不同的序列分别公示;将补充资料的审评队列按审评部门分别公示;将审评进程中的有关信息公开,包括各专业完成审评的信息、各主审报告完成审评任务的目标审评时间、与审评工作相关的会议信息等。
图3 药品审评中心组织机构

3.强化药物研究监督管理工作
截至目前,全国共有356家医疗机构的2438个临床专业通过GCP认证。2011年,国家食品药品监督管理局会同卫生部组织完成了对134家药物临床试验机构资格认定复核检查工作,对51家存在问题的机构或专业,责令整改或取消资格,有力地推动了药物临床试验质量规范的实施。组织召开了全国药物临床试验质量管理工作会议,明确了监管思路和工作方向,拟在现有管理基础上实施分类指导、重点培育,使一批条件较好的药物临床试验机构脱颖而出,承担起创新药物研究的重任,形成以探索性研究的临床试验机构为引领,以验证性研究的临床试验机构为基础的专业化、网络化的新格局,有力支持重大新药创制“十二五”规划的实施。
截至目前,全国共有46家非临床安全性评价研究机构通过《药物非临床研究质量管理规范(GLP)》认证。其中,有4家机构申请并通过了“经济合作与发展组织”(OECD)成员国的GLP检查,有2家机构接受了美国FDA的GLP检查,并获得通过,表明我国药品安全性评价能力得到国际认可,对于实现国家重大新药创制专项的目标,提升我国的新药研发水平,促进我国创新药物尽快走向国际市场具有重要意义。
各省级药品监管部门在开展注册现场核查中,不断规范和细化核查要求,严把申报资料质量。对一些申报资料不规范,质量不达标的药品注册申请,予以驳回或要求企业主动撤回。在生产现场检查中,加强对生产工艺可行性的检查,对打击研发中的弄虚作假行为、维护药品注册秩序发挥了重要作用。
4 . 首次开展进口药品境外生产现场检查
为加强对进口药品的监管,履行《药品管理法》赋予的职责,组织制定了进口药品境外生产现场检查工作程序和2011年进口药品境外现场检查试点工作方案。组成7个检查组,赴美国、法国、意大利、印度、匈牙利、韩国、日本等7个国家对贝伐珠单抗注射液、地诺前列素注射液、注射用盐酸吉西他滨等7个品种开展境外现场检查工作,实现了进口药品境外检查“零”的突破,充分展现了中国药品监管“为国把关、为民尽责”的信心和实力,也为加强监管能力建设、开展国际监管合作奠定了基础。
六、工作展望
近年来,随着医药产业的高速发展和新药研发能力的快速提高,药品注册管理工作不能充分满足社会各界的期望和要求,存在着服务能力与创新需求不相适应、审评审批策略与鼓励创新政策不相适应、体制机制与提高质量和效率的目标不相适应、药品质量标准水平与公众期望不相适应等问题。2012年,国家食品药品监督管理局将根据《国家药品安全“十二五”规划》的总体部署,做好以下三方面工作:
(一)构建科学的药品注册管理体系,进一步提高药品审评审批的质量和效率。从我国目前实行的国家和省两级注册管理体制的实际情况出发,调整审评审批策略和人力资源配置,优化顶层设计和管理流程,加强临床试验监管,完善信息管理系统及决策管理机制,确保创新药和临床亟需仿制药能够按时限开展审评,形成国家和省职责分明、权责一致、相互配合、互为补充的工作格局。
一是调整审评策略,建立鼓励先进的政策导向。要确立优先审评领域,实施优先审评策略,将有限的审评审批力量向创新药、新药创制重大专项支持项目以及临床短缺及亟需仿制药等优先领域、重点项目倾斜,确保公众用药可及性。要探索建立药品上市价值评估制度,对已有多家生产且不具备上市价值的申报品种,研究限制政策和措施,引导企业理性申报。
二是调整药品注册管理的事权。合理划分中央与地方,行政审批与技术审评事权,充分利用省局的人力资源和技术能力,授权具有资质和条件的省局承担部分药品注册申请事项的审评审批工作,逐步建立中央集中审评和国家管控的区域性药品审评中心辅助审评相结合的管理模式。
(二)全面提高仿制药质量,确保公众用药安全。以提高与淘汰相结合,政府引导推动与企业主动作为相结合,全面统筹与重点推进相结合为总体思路,以基本药物和临床常用品种为重点,开展仿制药质量一致性评价工作,促进仿制药品质量的持续提高,推动产品不断升级。达到质量一致性要求的,将得到药品在招标采购、定价等方面的优惠政策;达不到要求的,将予以淘汰。
(三)加强基础建设,提升服务能力。大力加强队伍建设,建立一支思想过硬、作风顽强、技术精湛的涵盖审评、检查、检验的专业化和专职化的药品注册管理队伍。加快药品标准信息化建设,实现药品标准的查询、检索、发布、分析、研究等管理工作的自动化和网络化,提高标准管理工作的质量和效率。
希望社会各界继续关心和支持药品注册管理工作。
(摘自:SFDA网站 2012-09-27)
 京公网安备 11010802027110号
京公网安备 11010802027110号